
Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals?


{kind=link}
Abstract
:Simple Summary
Abstract
1. CARs: The Ultimate Tumour Killing Machines?
2. Switching on the Ignition with Oncolytic Viruses
3. Combination OV and T Cell Therapies: Driving CAR T to the Tumour
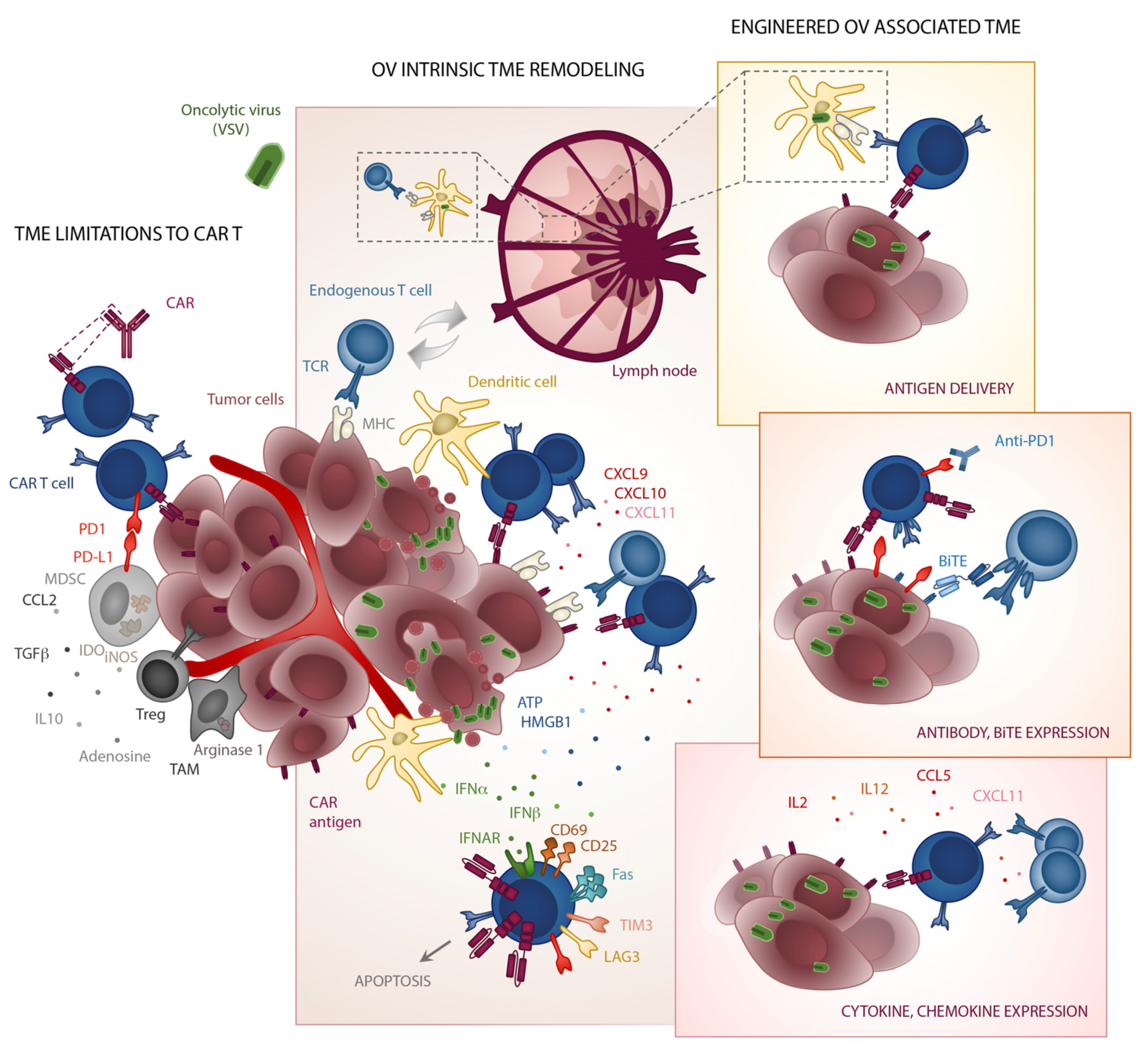
4. The TME—A Breaker’s Yard for CAR T Cells
5. TME Make over by Oncolytic Viruses
6. Fiddling while CARs Burn
7. Viruses as Micro-Pharmacies for T Cells
8. Graffitiing Antigenic Specificity onto Tumours
9. Virus CAR-Pooling to Tumours
10. Conclusions: OV Enhanced CAR T Cell Therapy—And Vice Versa
Funding
Acknowledgments
Conflicts of Interest
References
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2013, 257, 56–71. [Google Scholar] [CrossRef] [Green Version]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically Targeted T Cells Eradicate Systemic Acute Lymphoblastic Leukemia Xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2013, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef] [PubMed]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. OncoImmunology 2012, 1, 458–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; Mcgettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Hyrenius-Wittsten, A.; Roybal, K.T. Paving New Roads for CARs. Trends Cancer 2019, 5, 583–592. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density deter-mines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [Green Version]
- Au, G.G.; Lincz, L.F.; Enno, A.; Shafren, D.R. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br. J. Haematol. 2007, 137, 133–141. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B.; Gowrishankar, B.; Duraffour, S.; Andrei, G.; Upton, C.; Evans, D.H. Vaccinia Virus–Encoded Ribonucleotide Reductase Subunits Are Differentially Required for Replication and Pathogenesis. PLoS Pathog. 2010, 6, e1000984. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Rabkin, S.D. Designing herpes viruses as oncolytics. Mol. Ther. Oncolytics 2015, 2, 15010. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.-S.; Falls, T.; Burns, J.; Garcia, V.; et al. The Oncolytic Poxvirus JX-594 Selectively Replicates in and Destroys Cancer Cells Driven by Genetic Pathways Commonly Activated in Cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.T.; Aguirre-Hernández, C.; Halldén, G.; Parker, A.L. Designer Oncolytic Adenovirus: Coming of Age. Cancers 2018, 10, 201. [Google Scholar] [CrossRef] [Green Version]
- Ilkow, C.S.; Swift, S.L.; Bell, J.C.; Diallo, J.-S. From Scourge to Cure: Tumour-Selective Viral Pathogenesis as a New Strategy against Cancer. PLoS Pathog. 2014, 10, e1003836. [Google Scholar] [CrossRef] [Green Version]
- Pikor, L.A.; Bell, J.C.; Diallo, J.-S. Oncolytic Viruses: Exploiting Cancer’s Deal with the Devil. Trends Cancer 2015, 1, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.-Y.; Chen, S.; Zhao, X.; Lei, C.-Q.; Guo, L.; Chen, Y.; et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 2015, 17, 241–249. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; Tenoever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor selective replication of Newcastle disease virus: Association with defects of tumor cells in antiviral defence. Int. J. Cancer 2006, 119, 328–338. [Google Scholar] [CrossRef]
- Ahmed, M.; Cramer, S.D.; Lyles, D.S. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 2004, 330, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Obuchi, M.; Fernandez, M.; Barber, G.N. Development of recombinant vesicular stomatitis vi-ruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 2003, 77, 8843–8856. [Google Scholar] [CrossRef] [Green Version]
- Willmon, C.L.; Saloura, V.; Fridlender, Z.G.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Kottke, T.; Federspiel, M.; Barber, G.; Vile, R.G.; et al. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicu-lar stomatitis virus for therapy of mesothelioma. Cancer Res. 2009, 69, 7713–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Shaw, A.R.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Restifo, N.P. Cellular Constituents of Immune Escape within the Tumor Microenvironment: Figure 1. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, I.M.; Slingluff, C.L.; Lee, J.K.; Garbee, C.F.; Shu, J.; Anderson, S.G.; Mayer, M.E.; Knaus, W.A.; Mullins, D.W. CXC Chemokine Receptor 3 Expression by Activated CD8+ T cells Is Associated with Survival in Melanoma Patients with Stage III Disease. Cancer Res. 2004, 64, 7697–7701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, R.B.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Buchlis, G.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Crisanti, M.C.; Wang, L.-C.S.; Heitjan, D.; Snyder, L.A.; et al. CCL2 Blockade Augments Cancer Immunotherapy. Cancer Res. 2009, 70, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2+ Myeloid-Derived Suppressor Cells Promote Immune Escape by Limiting Activated CD8 T-cell Infiltration into the Tumor Microenvironment. Cancer Res. 2011, 72, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Toker, A.; Liu, Z.Q.; Ohashi, P.S. Turning the Tide against Regulatory T Cells. Front. Oncol. 2019, 9, 279. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. VSV Oncolytic Virotherapy in the B16 Model Depends Upon Intact MyD88 Signaling. Mol. Ther. 2011, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic Virotherapy and Immunogenic Cancer Cell Death: Sharpening the Sword for Improved Cancer Treatment Strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vloten, J.P.; Workenhe, S.T.; Wootton, S.K.; Mossman, K.L.; Bridle, B.W. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J. Immunol. 2018, 200, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, H.J.; Lee, W.S.; Yang, H.; Kong, S.J.; Lee, N.K.; Moon, E.S.; Choi, J.; Han, E.C.; Kim, J.H.; Ahn, J.B.; et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune checkpoint blockade. Clin. Cancer Res. 2018, 25, 1612–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, N.M.; Mulgrew, K.; McGlinchey, K.; Monks, N.R.; Ji, H.; Herbst, R.; Suzich, J.; Hammond, S.A.; Kelly, E.J. Oncolytic VSV Primes Differential Responses to Immuno-oncology Therapy. Mol. Ther. 2017, 25, 1917–1932. [Google Scholar] [CrossRef]
- Katayama, Y.; Tachibana, M.; Kurisu, N.; Oya, Y.; Terasawa, Y.; Goda, H.; Kobiyama, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H.; et al. Oncolytic Reovirus Inhibits Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a TLR3-Dependent Manner. J. Immunol. 2018, 200, 2987–2999. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 Checkpoint Blockade Enhances Oncolytic Measles Virus Therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [Green Version]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Patnaik, M.M.; Ruiz, A.; Russell, S.J.; Peng, K.-W. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood 2016, 127, 1449–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.-M.; Pandha, H.; Harrington, K.; et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti–PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2016, 19, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [Green Version]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Packiriswamy, N.; Upreti, D.; Zhou, Y.; Khan, R.; Miller, A.; Diaz, R.M.; Rooney, C.M.; Dispenzieri, A.; Peng, K.-W.; Russell, S.J. Oncolytic measles virus therapy enhances tumor antigen-specific T-cell responses in patients with multiple myeloma. Leukemia 2020, 34, 3310–3322. [Google Scholar] [CrossRef] [Green Version]
- Keppler, S.J.; Rosenits, K.; Koegl, T.; Vucikuja, S.; Aichele, P. Signal 3 Cytokines as Modulators of Primary Immune Responses during Infections: The Interplay of Type I IFN and IL-12 in CD8 T Cell Responses. PLoS ONE 2012, 7, e40865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, K.; Kim, S.-K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Selin, L.K.; Welsh, R.M. IFN-Induced Attrition of CD8 T Cells in the Presence or Absence of Cognate Antigen during the Early Stages of Viral Infections. J. Immunol. 2006, 176, 4284–4295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, K.; Hübner, A.; Davis, R.J.; Welsh, R.M. Analysis of Apoptosis of Memory T Cells and Dendritic Cells during the Early Stages of Viral Infection or Exposure to Toll-Like Receptor Agonists. J. Virol. 2010, 84, 6262. [Google Scholar] [CrossRef] [Green Version]
- Jangalwe, S.; Kapoor, V.N.; Xu, J.; Girnius, N.; Kennedy, N.J.; Edwards, Y.J.K.; Welsh, R.M.; Davis, R.J.; Brehm, M.A. Cutting Edge: Early Attrition of Memory T Cells during Inflammation and Costimulation Blockade Is Regulated Concurrently by Proapoptotic Proteins Fas and Bim. J. Immunol. 2019, 202, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, R.M.; Bahl, K.; Marshall, H.D.; Urban, S.L. Type 1 Interferons and Antiviral CD8 T-Cell Responses. PLoS Pathog. 2012, 8, e1002352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U.; Vivier, E.; Jonjic, S.; Oxenius, A. Type I Interferons Protect T Cells against NK Cell Attack Mediated by the Activating Receptor NCR1. Immunology 2014, 40, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Köhrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I Interferon Protects Antiviral CD8+ T Cells from NK Cell Cytotoxicity. Immunology 2014, 40, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Arulanandam, R.; Batenchuk, C.; Angarita, F.A.; Ottolino-Perry, K.; Cousineau, S.; Mottashed, A.; Burgess, E.; Falls, T.J.; De Silva, N.; Tsang, J.; et al. VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection. Cancer Cell 2015, 28, 210–224. [Google Scholar] [CrossRef] [Green Version]
- Kottke, T.; Hall, G.; Pulido, J.; Diaz, R.M.; Thompson, J.; Chong, H.; Selby, P.; Coffey, M.; Pandha, H.; Chester, J.; et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J. Clin. Investig. 2010, 120, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbach, C.J.; De Silva, N.S.; Falls, T.J.; Aladl, U.; Evgin, L.; Paterson, J.M.; Sun, Y.Y.; Roy, D.G.; Rintoul, J.L.; Daneshmand, M.; et al. Targeting Tumor Vasculature With an Oncolytic Virus. Mol. Ther. 2011, 19, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.-H.; et al. Oncolytic Vaccinia Virus Disrupts Tumor-Associated Vasculature in Humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Chen, H.; Rojas, J.; Sampath, P.; Thorne, S.H. Oncolytic vaccinia virus demonstrates antiangiogenic effects mediated by targeting of VEGF. Int. J. Cancer 2014, 135, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Matuszewska, K.; Santry, L.A.; Van Vloten, J.P.; Auyeung, A.W.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin. Cancer Res. 2018, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor–Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Wang, L.-C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology 2017, 7, e1395997. [Google Scholar] [CrossRef]
- Tanoue, K.; Shaw, A.R.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus–Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Aalipour, A.; Le Boeuf, F.; Tang, M.; Murty, S.; Simonetta, F.; Lozano, A.X.; Shaffer, T.M.; Bell, J.C.; Gambhir, S.S. Viral Delivery of CAR Targets to Solid Tumors Enables Effective Cell Therapy. Mol. Ther. Oncolytics 2020, 17, 232–240. [Google Scholar] [CrossRef]
- Park, A.K.; Fong, Y.; Kim, S.-I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.-C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-Modified T-Cell Therapy Cures Mice with Established Intracerebral Glioma and Generates Host Immunity against Tumor-Antigen Loss. Clin. Cancer Res. 2013, 20, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic Immunovirotherapy for Melanoma Using Vesicular Stomatitis Virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottke, T.; Errington, F.; Pulido, J.; Galivo, F.; Thompson, J.; Wongthida, P.; Diaz, R.M.; Chong, H.; Ilett, E.; Vile, R.; et al. Broad antigenic coverage induced by viral cDNA library-based vaccination cures established tumors. Nat. Med. 2011, 17, 854–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulido, J.; Kottke, T.; Thompson, J.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Rommelfanger, D.; Ilett, E.; Pease, L.; Pandha, H.; et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat. Biotechnol. 2012, 30, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating Cancer Immunotherapy Using an Oncolytic Virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Rességuier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba Virus as a Potent Oncolytic Vaccine Vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, M.J.; Stephenson, K.B.; Pol, J.; Wang, F.; Lefebvre, C.; Stojdl, D.F.; Nikota, J.K.; Dvorkin-Gheva, A.; Nguyen, A.; Chen, L.; et al. Customized Viral Immunotherapy for HPV-Associated Cancer. Cancer Immunol. Res. 2017, 5, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Overwijk, W.W.; Theoret, M.R.; Finkelstein, S.E.; Surman, D.R.; De Jong, L.A.; Vyth-Dreese, F.A.; Dellemijn, T.A.; Antony, P.A.; Spiess, P.J.; Palmer, D.C.; et al. Tumor Regression and Autoimmunity after Reversal of a Functionally Tolerant State of Self-reactive CD8+ T Cells. J. Exp. Med. 2003, 198, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Wongthida, P.; Diaz, R.M.; Pulido, C.; Rommelfanger, D.; Galivo, F.; Kaluza, K.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. Activating Systemic T-Cell Immunity Against Self Tumor Antigens to Support Oncolytic Virotherapy with Vesicular Stomatitis Virus. Hum. Gene Ther. 2011, 22, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.R.; Simovic, B.; Chen, L.; Bastin, D.; Nguyen, A.; Stephenson, K.; Mandur, T.S.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Endogenous T cells prevent tumor immune escape following adoptive T cell therapy. J. Clin. Investig. 2019, 129, 5400–5410. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Von Scheidt, B.; Davenport, A.J.; Beavis, P.A.; Westwood, J.A.; Mardiana, S.; Tscharke, D.C.; Ellis, S.; Prince, H.M.; Trapani, J.A.; et al. Dual-specific Chimeric Antigen Receptor T Cells and an Indirect Vaccine Eradicate a Variety of Large Solid Tumors in an Immunocompetent, Self-antigen Setting. Clin. Cancer Res. 2016, 23, 2478–2490. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer Res. 2019, 25, 7340–7350. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.-T. T-cell Engager-armed Oncolytic Vaccinia Virus Significantly Enhances Antitumor Therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speck, T.; Heidbuechel, J.P.; Veinalde, R.; Jaeger, D.; Von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy against Solid Tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus–Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Qiao, J.; Kottke, T.; Willmon, C.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Thompson, J.R.; Ryno, P.; Barber, G.N.; Chester, J.D.; et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat. Med. 2007, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Wang, H.; Kottke, T.; Diaz, R.M.; Willmon, C.L.; Hudacek, A.W.; Thompson, J.R.; Parato, K.; Bell, J.C.; Naik, J.D.; et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008, 15, 604–616. [Google Scholar] [CrossRef]
- Kottke, T.; Diaz, R.M.; Kaluza, K.; Pulido, J.; Galivo, F.; Wongthida, P.; Thompson, J.; Willmon, C.; Barber, G.N.; Chester, J.; et al. Use of Biological Therapy to Enhance Both Virotherapy and Adoptive T-Cell Therapy for Cancer. Mol. Ther. 2008, 16, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic Antitumor Effects of Immune Cell-Viral Biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef]
- Thorne, S.H.; Liang, W.; Sampath, P.; Schmidt, T.; Sikorski, R.; Beilhack, A.; Contag, C.H. Targeting Localized Immune Suppression Within the Tumor Through Repeat Cycles of Immune Cell-oncolytic Virus Combination Therapy. Mol. Ther. 2010, 18, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- VanSeggelen, H.; Tantalo, D.G.; Afsahi, A.; Hammill, J.A.; Bramson, J.L. Chimeric antigen receptor–engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics 2015, 2, 15014. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.P.; Farrar, J.D. Regulation of effector and memory T-cell functions by type I interferon. Immunology 2011, 132, 466–474. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evgin, L.; Vile, R.G. Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals? Cancers 2021, 13, 1106. https://doi.org/10.3390/cancers13051106
Evgin L, Vile RG. Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals? Cancers. 2021; 13(5):1106. https://doi.org/10.3390/cancers13051106
Chicago/Turabian StyleEvgin, Laura, and Richard G. Vile. 2021. "Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals?" Cancers 13, no. 5: 1106. https://doi.org/10.3390/cancers13051106
APA StyleEvgin, L., & Vile, R. G. (2021). Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals? Cancers, 13(5), 1106. https://doi.org/10.3390/cancers13051106