
Signaling Pathways That Control Apoptosis in Prostate Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
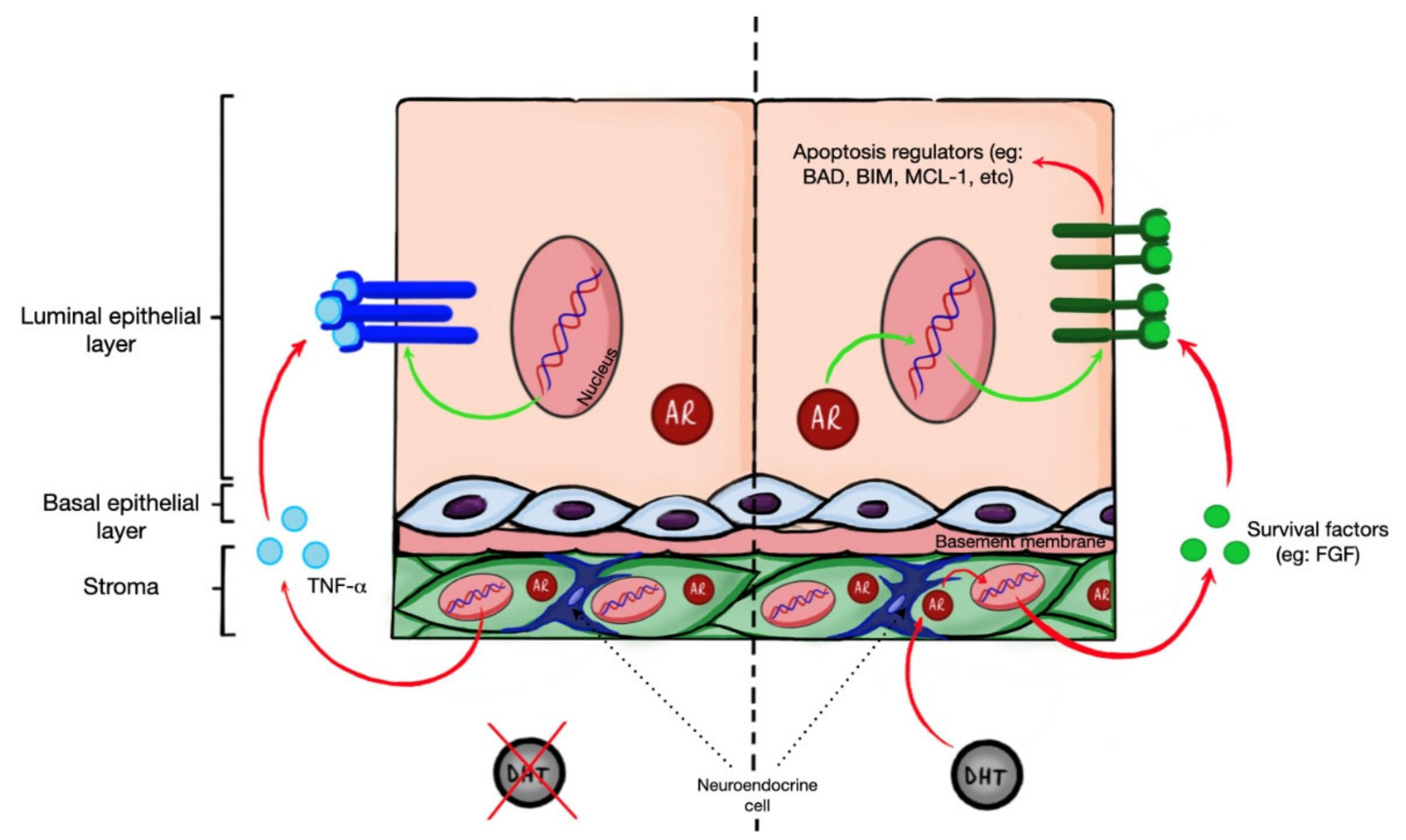
2. Apoptosis in Prostate Epithelial Cells Is Controlled by AR Signaling in Stroma
2.1. Stroma and Luminal Compartments of the Prostate Gland
2.2. Androgen Ablation Triggers Apoptosis
2.3. Role of Stromal AR in Epithelial Apoptosis Regulation
2.4. Roles of Mitochondrial and Death Receptor-Induced Apoptosis in Prostate Involution
3. BCL-2 Family Proteins in Prostate Cancer
3.1. Anti-Apoptotic Proteins
3.2. Pro-Apoptotic Effector Proteins
3.3. BH3-Only Proteins
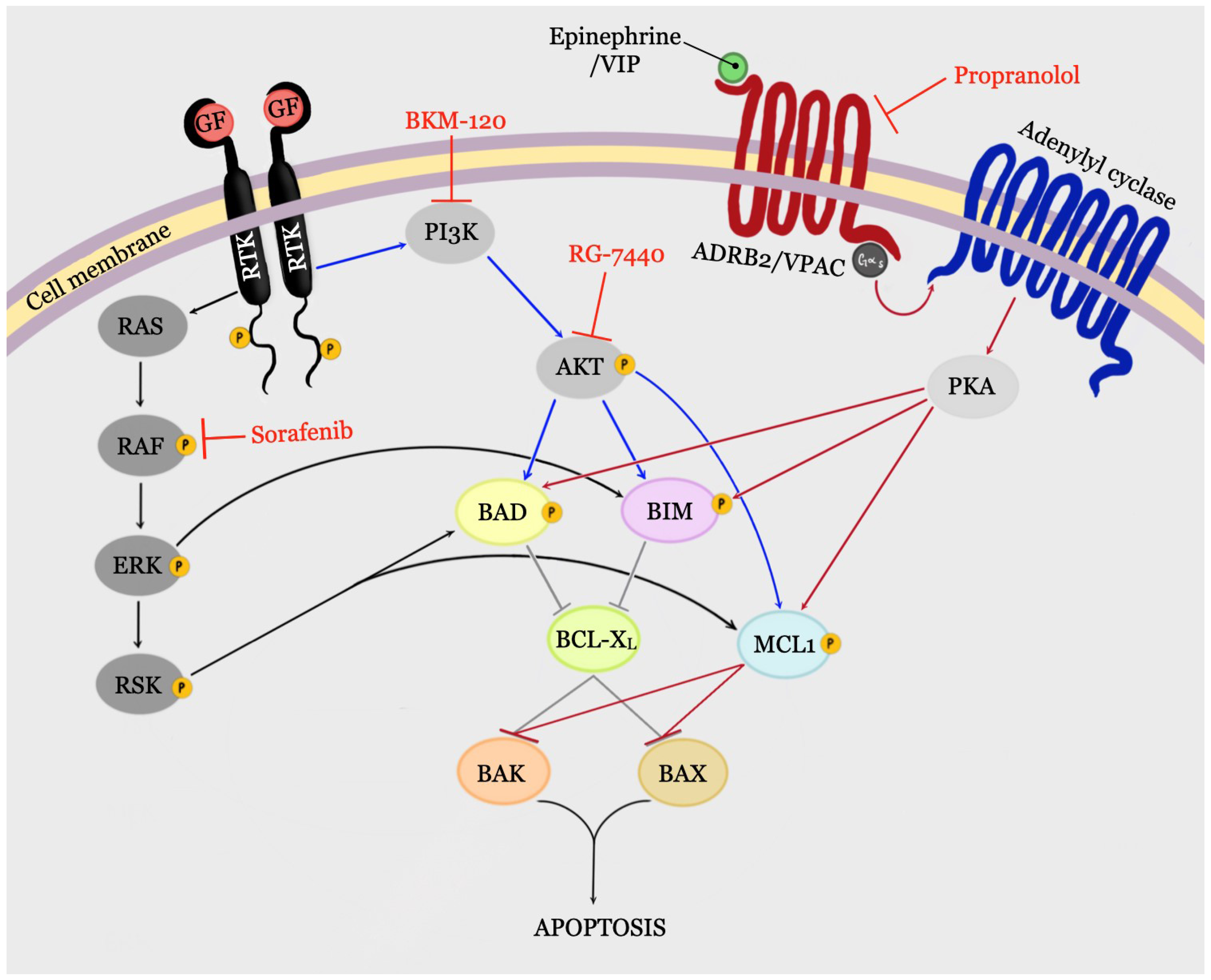
4. Signaling Pathways That Can Control BCL-2 Family Proteins
4.1. PI3K/AKT Signaling Pathway
4.2. RAS/ERK Signaling Pathway
4.3. GPCR/PKA Signaling
4.3.1. VIP/PKA Signaling
4.3.2. ADRB2/PKA Signaling
5. Clinical Trials of Drugs Targeting Anti-Apoptotic Signaling in PCa

{kind=link}
{kind=link}
| Agent | Target(s) | Population | Phase | Outcome |
|---|---|---|---|---|
| Ipatasertib (GDC-0068, RG-7440) + abiraterone | All AKT isoforms + AR | PTEN-negative mCRPC | Phase II | • Improved rPFS (8.18 mo (400 mg ipatasertib) and 8.31 mo (200 mg ipatasertib) vs. 6.37 mo for placebo) [212] |
| MLN0128 (Sapanisertib, INK 128) | TORC1 and TORC2 | mCRPC—after abiraterone acetate and/or enzalutamide | Phase II | • PSA rise on treatment in all patients (median, 159% increase from baseline); PSA declined immediately in 4 patients upon drug discontinuation [215] |
| Temsirolimus (Torisel, CCI-779) | MTOR | mCRPC | Phase II |
|
| Everolimus (RAD001, Afinitor, Zortress) | MTOR | chemotherapy-naïve mCRPC patients | Phase II |
|
| Everolimus + bicalutamide | MTOR +AR | CRPC—previously treated with bicalutamide | Phase II |
|
| Bicalutamide-naïve CRPC | Phase II | • ≥30% PSA decline in 75% of patients [223]. | ||
| Everolimus + carboplatin + prednisone | MTOR + DNA replication + GR | mCRPC pretreated with docetaxel | Phase II |
|
| Buparlisib (BKM-120) + enzalutamide | Class I PI3K + AR | mCRPC progressing on or are not candidates for docetaxel | Phase II |
|
| PX-866 +/− abiraterone acetate | Class I PI3K + AR | recurrent or metastatic CRPC | Phase II |
|
| Temsirolimus (Torisel, CCI-779) | MTOR | Docetaxel-treated CRPC patients | Phase II |
|
| Chemotherapy-naïve CRPC |
| |||
| Ridaforolimus (AP23573 MK8669) | MTOR | Taxane-treated CRPC patients | Phase II |
|
| Ridaforolimus + bicalutamide | MTOR + AR | AsymptomaticmCRPC patients | Phase II |
|
| Everolimus + gefitinib (Iressa, ZD 1839) | MTOR + EGFR | CRPC | Phase I/II |
|
| cabozantinib (Cabometyx, Cometriq, Cabozanix, BMS-907351, XL184) | RTK | advanced, recurrent or metastatic cancers | Phase II | • CRPC patients had largest PFS (median, 5.5 mo vs. 1.4 mo for placebo) [228] |
| CRPC | Phase II | • Improved PFS (median, 23.9 weeks with cabozantinib vs. 5.9 weeks with placebo) [227]. | ||
| ||||
| mCRPC—previously treated with docetaxel and abiraterone and/or enzalutamide | Phase III |
| ||
| Sunitinib (Sutent, SU11248, SU011248) | RTK | CRPC | Phase II |
|
| Afatinib (Gilotrif, BIBW 2992 MA2) | EGFR | CRPC | Phase II | • limited anti-tumor activity [232] |
| Gefitinib (ZD1839, Iressa) | EGFR | mCRPC progressing on LHRH analog + antiandrogen (bicalutamide or flutamide). | Phase II | • No PSA or objective response in any patient [236]. |
| nmCRPC | Phase II | • None of the evaluable patients had a PSA response [238]. | ||
| Chemotherapy-naïve CRPC | Phase II |
| ||
| CRPC | Phase II |
| ||
| Gefitinib + docetaxel | EGFR + tubulin | CRPC | Phase II | • Response rate and duration were consistent with those of docetaxel monotherapy [237] |
| Gefitinib + prednisone | EGFR + GR | CRPC | Phase II |
|
| Gefitinib + antiandrogen + LH-RH analogue | EGFR + AR + GnRH receptor | CRPC | Phase II | • Gefitinib treatment did not result in any objective response (PSA or OR). Median time to progression was 70 days. Median OS was 293 days [235]. |
| Vandetanib (ZACTIMA) + bicalutamide | VEGFR2/EGFR + AR | Chemotherapy-naïve mCRPC (rising PSA on ADT and minimally symptomatic) | Phase II |
|
| Vandetanib | VEGFR2/EGFR | mCRPC | Phase II |
|
| cetuximab | EGFR | mCRPC | Phase Ib/IIa |
|
| cetuximab + mitoxantrone + prednisone | EGFR + topoisomerase II + GR | mCRPC progressing after docetaxel | Phase II | • Median time to progression (TTP): 4.9 mo (cetuximab + mitoxantrone + prednisone) and 6.6 mo (mitoxantrone + prednisone); measurable disease response rate: 2% and 4%; PSA response rate: 7.7% and 17.6%; median survival: 11.9 and 15.7 mo, respectively [249]. |
| cetuximab + docetaxel | EGFR+tubulin | mCRPC | Phase II |
|
| Pertuzumab | HER2 dimerization | CRPC progressing after at least one taxane-based regimen | Phase II |
|
| Lapatinib (Tykerb, GSK572016, GW2016, GW-572016) | EGFR and HER2 | Chemotherapy-naïve CRPC patients with rising PSA on ADT | Phase II |
|
| Erlotinib (Tarceva, CP-358,774, CP358774, OSI774, OSI-774) | EGFR | Advanced or metastatic PCa (including CRPC) | Phase II |
|
| chemotherapy-naive CRPC | Phase II |
| ||
| Erlotinib + docetaxel | EGFR + tubulin | ≥ 65 years CRPC patients progressing despite ADT | Phase II |
|
| Trastuzumab (Herceptin) | HER2 | CRPC | Phase II |
|
| Trastuzumab + docetaxel | HER2 + tubulin | HER2-positive CRPC |
| |
| Cediranib | VEGFR | mCRPC—progressing following docetaxel therapy | Phase II | • 6/39 patients had confirmed PR; 1 had an unconfirmed PR. 43.9% of patients were progression-free at 6 mo; for all patients, median PFS: 3.7 mo; OS: 10.1 mo [260] |
| Cediranib + DP | VEGFR | mCRPC | Phase II | • 6-mo PFS rate: 61% in DP+C arm and 57% in DP arm. There were no significant differences in 6-mo OS rate, objective tumor and PSA response rates, and biomarkers in the two arms [261]. |
| Bevacizumab + temsirolimus | VEGF-A & MTOR | mCRPC—previously treated with chemotherapy | Phase I-II | • Median time to progression: 2.6 mo; median best PSA change: 32% increase (met the predefined futility rule leading to early termination of the study) [262] |
| Bevacizumab + DP | VEGF-A | mCRPC | Phase III | • Improved PFS (median, 9.9 mo for DP+B vs. 7.5 mo for DP) and OR (49.4% vs. 35.5%;) but not OS (median, 22.6 mo vs. 21.5 mo) [255]. |
| bevacizumab + docetaxel + estramustine | VEGF-A + tubulin + MAPs and tubulin | CRPC | Phase II |
|
| Dovitinib (CHIR-258, TKI258) | VEGFR, FGFR, PDGFR | mCRPC (80% were post-docetaxel) | Phase II |
|
| Pazopanib (Votrient, GW786034B) + bicalutamide | VEGFR + AR | chemotherapy-naive (CRPC). | Phase II |
|
| Aflibercept (Eylea, Zaltrap) + Docetaxel + prednisone | VEGFA/B & PGF + tubulin + GR | Chemotherapy-naïve mCRPC | Phase III | • Median OS: 22·1 mo (aflibercept + DP) and 21·2 mo (DP) no clinical benefit compared to control group [266]. |
| SU5416 + dexamethasone | VEGFR2 | Chemotherapy-naïve CRPC | Phase II | • No detectable effect on PSA secretion or time to progression [267]. |
| Thalidomide (Contergan, Thalomid, Talidex) | pro-angiogenic factors (e.g., VEGF, BFGF, IL-6 | mCRPC (failed other therapy forms) | Phase II | • ≥50% PSA decline in 18% of patients (200 mg/day) but no decline in the high-dose arm (200 mg/day to 1200 mg/day); PSA decline was maintained for >150 days in 4 patients; 28% of all patients had >40% PSA decline; 13% had 40% to 50% PSA decline [268]. |
| ||||
| Itraconazole | Angiogenesis Hedgehog signaling | chemotherapy-naïve mCRPC | Phase II | • PSA PFS rates at 24 weeks: 11.8% in 200 mg/day arm and 48.0% in the 600 mg/day arm; median PFS: 11.9 weeks and 35.9 weeks, PSA response rates: 0% and 14.3%, respectively [270]. |
| Sorafenib | RAF VEGFR2 PDGFRB | mCRPC | Phase II |
|
| ||||
| ISIS 5132 | RAF1 | chemotherapy-naïve, mCRPC | Phase II |
|
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Alghamidi, I.G.; Hussain, I.I.; Alghamdi, M.S.; El-Sheemy, M.A. The Incidence Rate of Prostate Cancer in Saudi Arabia: An Observational Descriptive Epidemiological Analysis of Data from the Saudi Cancer Registry 2001–2008. Hematol. Oncol. Stem Cell Ther. 2014, 7, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdana, N.R.; Mochtar, C.A.; Umbas, R. The Risk Factors of Prostate Cancer and Its Prevention: A Literature Review. Acta Med. Indones. 2016, 48, 11. [Google Scholar]
- Schrecengost, R.S.; Knudsen, K.E. Molecular Pathogenesis and Progression of Prostate Cancer. Semin. Oncol. 2013, 40, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Apata, T.; Gordetsky, J.B.; Singh, R. Docetaxel Combined with Thymoquinone Induces Apoptosis in Prostate Cancer Cells via Inhibition of the PI3K/AKT Signaling Pathway. Cancers 2019, 11, 1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattrini, C.; Castro, E.; Lozano, R.; Zanardi, E.; Rubagotti, A.; Boccardo, F.; Olmos, D. Current Treatment Options for Metastatic Hormone-Sensitive Prostate Cancer. Cancers 2019, 11, 1355. [Google Scholar] [CrossRef] [Green Version]
- Green, S.M.; Mostaghel, E.A.; Nelson, P.S. Androgen Action and Metabolism in Prostate Cancer. Mol. Cell. Endocrinol. 2012, 360, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Daneshmand, S.; Ahmadi, H. Androgen Deprivation Therapy for Prostate Cancer: Long-Term Safety and Patient Outcomes. Patient Relat. Outcome Meas. 2014, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Dutt, S.S.; Gao, A.C. Molecular Mechanisms of Castration-Resistant Prostate Cancer Progression. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liao, C.-Y.; Chtatou, H.; Incrocci, L.; van Gent, D.C.; van Weerden, W.M.; Nonnekens, J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers 2019, 11, 1593. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.; Edlind, M. PI3K-AKT-MTOR Signaling in Prostate Cancer Progression and Androgen Deprivation Therapy Resistance. Asian J. Androl. 2014, 16, 378. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Gleave, M.E. Co-targeting Driver Pathways in Prostate Cancer: Two Birds with One Stone. EMBO Mol. Med. 2018, 10, e8928. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.-M.; Pramanik, R.; Nicholson, L.J.; Dew, T.K.; Martin, F.L.; Muir, G.H.; Morris, J.D.H. Ras-MEK-ERK Signaling Cascade Regulates Androgen Receptor Element-Inducible Gene Transcription and DNA Synthesis in Prostate Cancer Cells. Int. J. Cancer 2007, 121, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-Targeted Therapy in Men with Prostate Cancer: Evolving Practice and Future Considerations. Prostate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil Da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, F.; Sharma, N.V.; Moran, J.D.; Moreno, C.S. The Biology of Castration-Resistant Prostate Cancer. Curr. Probl. Cancer 2015, 39, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. The Context of Prostate Cancer Genomics in Personalized Medicine. Oncol. Lett. 2017, 13, 3347–3353. [Google Scholar] [CrossRef] [Green Version]
- Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.R.; Searle, J. Deletion of Cells by Apoptosis during Castration-Induced Involution of the Rat Prostate. Virchows Arch. B Cell Pathol 1973, 13, 87–102. [Google Scholar] [CrossRef]
- Kyprianou, N.; Isaacs, J.T. Activation of Programmed Cell Death in the Rat Ventral Prostate after Castration. Endocrinology 1988, 122, 552–562. [Google Scholar] [CrossRef]
- Toivanen, R.; Shen, M.M. Prostate Organogenesis: Tissue Induction, Hormonal Regulation and Cell Type Specification. Development 2017, 144, 1382–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velcheti, V.; Karnik, S.; Bardot, S.F.; Prakash, O. Pathogenesis of Prostate Cancer: Lessons from Basic Research. Ochsner J. 2008, 8, 213–218. [Google Scholar]
- Debes, J.D.; Tindall, D.J. The Role of Androgens and the Androgen Receptor in Prostate Cancer. Cancer Lett. 2002, 187, 1–7. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2019, 37, 288. [Google Scholar] [CrossRef] [PubMed]
- Regter, S.; Hedayati, M.; Zhang, Y.; Zhou, H.; Dalrymple, S.; Koch, C.J.; Isaacs, J.T.; DeWeese, T.L. Androgen Withdrawal Fails to Induce Detectable Tissue Hypoxia in the Rat Prostate: Undetectable Tissue Hypoxia in the Rat Prostate. Prostate 2014, 74, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, C.; Clark, P.J. Quantitative Studies of Prostatic Secretion II. The Effect of Castration and of Estrogen Injection on the Normal and on the Hyperplastic Prostate Glands of Dogs. J. Exp. Med. 1940, 747–762. [Google Scholar] [CrossRef] [Green Version]
- Helminen, H.J.; Ericsson, J.L.E. Ultrastructural Studies on Prostatic Involution in the Rat Changes in the Secretory Pathways. J. Ultrastruct. Res. 1972, 40, 152–166. [Google Scholar] [CrossRef]
- Sandford, N.L.; Searle, J.W.; Kerr, J.F.R. Successive Waves of Apoptosis in the Rat Prostate after Repeated Withdrawal of Testosterone Stimulation. Pathology 1984, 16, 406–410. [Google Scholar] [CrossRef]
- English, H.F.; Santen, R.J.; Lsaacs, J.T. Response of Glandular versus Basal Rat Ventral Prostatic Epithelial Cells to Androgen Withdrawal and Replacement. Prostate 1987, 11, 229–242. [Google Scholar] [CrossRef]
- Bruckheimer, E.M.; Cho, S.; Brisbay, S.; Johnson, D.J.; Greenberg, N.; McDonnell, T.J. The Impact of Bcl-2 Expression and Bax Defciency on Prostate Homeostasis. Oncogene 2000, 19, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Kurita, T.; Wang, Y.Z.; Donjacour, A.A.; Zhao, C.; Lydon, J.P.; O’Malley, B.W.; Isaacs, J.T.; Dahiya, R.; Cunha, G.R. Paracrine Regulation of Apoptosis by Steroid Hormones in the Male and Female Reproductive System. Cell Death Differ. 2001, 8, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative Potential of Prostate Luminal Cells Revealed by Single-Cell Analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Wang, J.; Shang, Z.; Huang, S.-P.; Shyr, C.-R.; Yeh, S.; Chang, C. Increased CK5/CK8-Positive Intermediate Cells with Stromal Smooth Muscle Cell Atrophy in the Mice Lacking Prostate Epithelial Androgen Receptor. PLoS ONE 2011, 6, e20202. [Google Scholar] [CrossRef]
- Wu, C.-T.; Altuwaijri, S.; Ricke, W.A.; Huang, S.-P.; Yeh, S.; Zhang, C.; Niu, Y.; Tsai, M.-Y.; Chang, C. Increased Prostate Cell Proliferation and Loss of Cell Differentiation in Mice Lacking Prostate Epithelial Androgen Receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 12679–12684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Altuwaijri, S.; Lai, K.-P.; Wu, C.-T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen Receptor Is a Tumor Suppressor and Proliferator in Prostate Cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanbrough, M.; Leav, I.; Kwan, P.W.L.; Bubley, G.J.; Balk, S.P. Prostatic Intraepithelial Neoplasia in Mice Expressing an Androgen Receptor Transgene in Prostate Epithelium. Proc. Natl. Acad. Sci. USA 2001, 10823–10828. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhang, C.; Lin, C.-C.; Niu, Y.; Lai, K.-P.; Chang, H.; Yeh, S.-D.; Chang, C.; Yeh, S. Altered Prostate Epithelial Development and IGF-1 Signal in Mice Lacking the Androgen Receptor in Stromal Smooth Muscle Cells. Prostate 2011, 71, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, M.; Moffat, L.; McNeilly, A.; Brownstein, D.; Saunders, P.T.K.; Sharpe, R.M.; Smith, L.B. Smooth Muscle Cell-Specific Knockout of Androgen Receptor: A New Model for Prostatic Disease. Endocrinology 2011, 152, 3541–3551. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yeh, C.-R.; Niu, Y.; Chang, H.-C.; Tsai, Y.-C.; Moses, H.L.; Shyr, C.-R.; Chang, C.; Yeh, S. Altered Prostate Epithelial Development in Mice Lacking the Androgen Receptor in Stromal Fibroblasts: Altered Prostate Epithelial Development in Mice. Prostate 2012, 72, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.-P.; Yamashita, S.; Vitkus, S.; Shyr, C.-R.; Yeh, S.; Chang, C. Suppressed Prostate Epithelial Development with Impaired Branching Morphogenesis in Mice Lacking Stromal Fibromuscular Androgen Receptor. Mol. Endocrinol. 2012, 26, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Chang, H.-C.; Tian, J.; Shang, Z.; Niu, Y.; Chang, C. Stromal Androgen Receptor Roles in the Development of Normal Prostate, Benign Prostate Hyperplasia, and Prostate Cancer. Am. J. Pathol. 2015, 185, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Liu, Y.; Cai, T.; Horton, C.; Stefanson, J.; Wang, Z.A. Dissecting Cell-Type-Specific Roles of Androgen Receptor in Prostate Homeostasis and Regeneration through Lineage Tracing. Nat. Commun. 2017, 8, 14284. [Google Scholar] [CrossRef] [Green Version]
- Godoy, A.; Montecinos, V.P.; Gray, D.R.; Sotomayor, P.; Yau, J.M.; Vethanayagam, R.R.; Singh, S.; Mohler, J.L.; Smith, G.J. Androgen Deprivation Induces Rapid Involution and Recovery of Human Prostate Vasculature. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E263–E275. [Google Scholar] [CrossRef] [Green Version]
- Lekås, E.; Johansson, M.; Widmark, A.; Bergh, A.; Damber, J.-E. Decrement of Blood Flow Precedes the Involution of the Ventral Prostate in the Rat after Castration. Urol. Res. 1997, 25, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Shabsigh, A.; Ghafar, M.A.; Anastasiadis, A.G.; Buttyan, R. Biomarker Analysis Demonstrates a Hypoxic Environment in the Castrated Rat Ventral Prostate Gland. J. Cell. Biochem. 2001, 81, 437–444. [Google Scholar] [CrossRef]
- Verbrugge, I.; Johnstone, R.W.; Smyth, M.J. SnapShot: Extrinsic Apoptosis Pathways. Cell 2010, 143, 1192–1192.e2. [Google Scholar] [CrossRef] [Green Version]
- Mongiat, M.; Ligresti, G.; Marastoni, S.; Lorenzon, E.; Doliana, R.; Colombatti, A. Regulation of the Extrinsic Apoptotic Pathway by the Extracellular Matrix Glycoprotein EMILIN2. Mol. Cell. Biol. 2007, 27, 7176–7187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, L.E.; Hahne, M.; Viard, I.; Radlgruber, G.; Zanone, R.; Becker, K.; Müller, C.; Tschopp, J. Fas and Fas Ligand in Embryos and Adult Mice: Ligand Expression in Several Immune-Privileged Tissues and Coexpression in Adult Tissues Characterized by Apoptotic Cell Turnover. J. Cell Biol. 1996, 133, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Xerri, L.; Devilard, E.; Hassoun, J.; Mawas, C.; Birg, F. Fas Ligand Is Not Only Expressed in Immune Privileged Human Organs but Is Also Coexpressed with Fas in Various Epithelial Tissues. Mol. Pathol. 1997, 50, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.-Y.; Rubin, M.A.; Omene, C.; Lederman, S.; Stein, C.A. Fas Ligand Is Constitutively Secreted by Prostate Cancer Cells in Vitro. Clin. Cancer Res. 1998, 4, 1803–1811. [Google Scholar] [PubMed]
- Hyer, M.L.; Voelkel-Johnson, C.; Rubinchik, S.; Dong, J.; Norris, J.S. Intracellular Fas Ligand Expression Causes Fas-Mediated Apoptosis in Human Prostate Cancer Cells Resistant to Monoclonal Antibody-Induced Apoptosis. Mol. Ther. 2000, 2, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Matsuzawa, A.; Iguchi, T. Down Regulation of Bcl-2 Is the First Step on Fas-Mediated Apoptosis of Male Reproductive Tract. Oncogene 1996, 13, 31–37. [Google Scholar]
- De la Taille, A.; Chen, M.W.; Shabsigh, A.; Bagiella, E.; Kiss, A.; Buttyan, R. Fas Antigen/CD-95 Upregulation and Activation during Castration-Induced Regression of the Rat Ventral Prostate Gland. Prostate 1999, 40, 89–96. [Google Scholar] [CrossRef]
- Gao, S.; Lee, P.; Wang, H.; Gerald, W.; Adler, M.; Zhang, L.; Wang, Y.-F.; Wang, Z. The Androgen Receptor Directly Targets the Cellular Fas/FasL-Associated Death Domain Protein-Like Inhibitory Protein Gene to Promote the Androgen-Independent Growth of Prostate Cancer Cells. Mol. Endocrinology 2005, 19, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raclaw, K.A.; Heemers, H.V.; Kidd, E.M.; Dehm, S.M.; Tindall, D.J. Induction of FLIP Expression by Androgens Protects Prostate Cancer Cells from TRAIL-Mediated Apoptosis. Prostate 2008, 68, 1696–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornforth, A.N.; Davis, J.S.; Khanifar, E.; Nastiuk, K.L.; Krolewski, J.J. FOXO3a Mediates the Androgen-Dependent Regulation of FLIP and Contributes to TRAIL-Induced Apoptosis of LNCaP Cells. Oncogene 2008, 27, 4422–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lu, J.; Tindall, D.J. Androgens Regulate TRAIL-Induced Cell Death in Prostate Cancer Cells via Multiple Mechanisms. Cancer Lett. 2013, 335, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, H.; Zhang, X.; Chen, M.W.; Walsh, K.; Buttyan, R. An Elevated Bax/Bcl-2 Ratio Corresponds with the Onset of Prostate Epithelial Cell Apoptosis. Cell Death Differ. 1999, 6, 48–54. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, M.W.; Ng, A.; Ng, P.Y.; Lee, C.; Rubin, M.; Olsson, C.A.; Buttyan, R. Abnormal Prostate Development in C3(1)-Bcl-2 Transgenic Mice. Prostate 1997, 32, 16–26. [Google Scholar] [CrossRef]
- Cui, J.; Placzek, W. Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. Int. J. Mol. Sci. 2018, 19, 308. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Ann. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R. Apoptotic Pathways: Paper Wraps Stone Blunts Scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 Arbiters of Apoptosis and Their Growing Role as Cancer Targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P. Tumor-Specific Induction of the Intrinsic Apoptotic Pathway—A New Therapeutic Option for Advanced Prostate Cancer? Front. Oncol. 2019, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Xu, L. Chemosensitization of Prostate Cancer by Modulating Bcl-2 Family Proteins. Curr. Drug Targets 2010, 11, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, K.S.; Abel, P.D.; Lalani, E.-N. Role of the BcI-2 Gene Family in Prostate Cancer Progression and Its Implications for Therapeutic Intervention. Environ. Health Perspect. 1999, 107, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Haldar, S.; Negrini, M.; Monne, M.; Sabbioni, S.; Croce, C.M. Down-Regulation of Bcl-2 by P53 in Breast Cancer Cells. Cancer Res. 1994, 54, 2095–2097. [Google Scholar]
- Loeb, D.M. WT1 Influences Apoptosis Through Transcriptional Regulation of Bcl-2 Family Members. Cell Cycle 2006, 5, 1249–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catz, S.D.; Johnson, J.L. Transcriptional Regulation of Bcl-2 by Nuclear Factor KB and Its Significance in Prostate Cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kelly, W.K.; Fu, A.; Haines, K.; Hoffman, A.; Zheng, T.; Zhu, Y. Genome-Wide Methylation Analysis Identifies Involvement of TNF-α Mediated Cancer Pathways in Prostate Cancer. Cancer Lett. 2011, 302, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Wang, P.; Yu, X.; Wang, A.; Chai, G.; Fan, Y.; Zhang, Z. Systems Analysis of Phosphorylation-Regulated Bcl-2 Interactions Establishes a Model to Reconcile the Controversy over the Significance of Bcl-2 Phosphorylation: Enhanced Anti-Apoptotic Ability of Phosphorylated Bcl-2. Br. J. Pharmacol. 2019, 176, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Hockenbery, D.M.; Zutter, M.; Hickey, W.; Nahm, M.; Korsmeyer, S.J. BCL2 Protein Is Topographically Restricted in Tissues Characterized by Apoptotic Cell Death. Proc. Natl. Acad. Sci. USA 1991, 88, 6961–6965. [Google Scholar] [CrossRef] [Green Version]
- Catz, S.D.; Johnson, J.L. BCL-2 in Prostate Cancer: A Minireview. Apoptosis 2003, 8, 29–37. [Google Scholar] [CrossRef]
- McDonnell, T.J.; Troncoso, P.; Brisbay, S.M.; Logothetis, C.; Chung, L.W.K.; Hsieh, J.-T.; Tu, S.-M.; Campbell, M.L. Expression of the Protooncogene Bcl-2 in the Prostate and Its Association with Emergence of Androgen-Independent Prostate Cancer. Cancer Res. 1992, 52, 6940–6944. [Google Scholar]
- Love, H.D.; Booton, S.E.; Boone, B.E.; Breyer, J.P.; Koyama, T.; Revelo, M.P.; Shappell, S.B.; Smith, J.R.; Hayward, S.W. Androgen Regulated Genes in Human Prostate Xenografts in Mice: Relation to BPH and Prostate Cancer. PLoS ONE 2009, 4, e8384. [Google Scholar] [CrossRef] [Green Version]
- Westin, P.; Stattin, P.; Damber, J.-E. Castration Therapy Rapidly Induces Apoptosis in a Minority and Decreases Cell Proliferation in a Majority of Human Prostatic Tumors. Am. J. Pathol. 1995, 146, 1368–1375. [Google Scholar]
- Symmans, F.; Gil, S.; Chopin, D.; Olsson, C.A.; Korsmeyer, S.; Buttyan, R. Detection of the Apoptosis-Suppressing Oncoprotein Bcl-2 in Hormone-Refractory Human Prostate Cancers. Am. J. Pathol. 1993, 143, 390–400. [Google Scholar]
- Li, Q.; Deng, Q.; Chao, H.-P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking Prostate Cancer Cell AR Heterogeneity to Distinct Castration and Enzalutamide Responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [Green Version]
- Bonkhoff, H.; Fixemer, T.; Remberger, K. Relation between Bcl-2, Cell Proliferation, and the Androgen Receptor Status in Prostate Tissue and Precursors of Prostate Cancer. Prostate 1998, 34, 251–258. [Google Scholar] [CrossRef]
- Raffo, A.J.; Perlman, H.; Chen, M.-W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of Bcl-2 Protects Prostate Cancer Cells from Apoptosis in Vitro and Confers Resistance to Androgen Depletion in Vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- Placzek, W.J.; Wei, J.; Kitada, S.; Zhai, D.; Reed, J.C.; Pellecchia, M. A Survey of the Anti-Apoptotic Bcl-2 Subfamily Expression in Cancer Types Provides a Platform to Predict the Efficacy of Bcl-2 Antagonists in Cancer Therapy. Cell Death Dis. 2010, 1, e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of Pp60c-Src Reduces Bcl-XL Expression and Reverses the Transformed Phenotype of Cells Overexpressing EGF and HER-2 Receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, J.M.; Zeng, X.-R.; Boise, L.H. Regulation of Bcl-XL: A Little Bit of This and a Little Bit of STAT. Curr. Opin. Oncol. 2000, 12, 543–549. [Google Scholar] [CrossRef]
- Boise, H.; GonzBlez-Garcia, M.; Postema, E.; Ding, L.; Turka, L.A.; NuAez, G. Bcl-x, a Bcl-2-Related Gene That Functions as a Dominant Regulator of Apoptotic Cell Death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Poruchynsky, M.S.; Wang, E.E.; Rudin, C.M.; Blagosklonny, M.V.; Fojo, T. Bcl-XL Is Phosphorylated in Malignant Cells Following Microtubule Disruption. Cancer Res. 1998, 58, 3331–3338. [Google Scholar]
- Clem, R.J.; Cheng, E.H.-Y.; Karp, C.L.; Kirsch, D.G.; Ueno, K.; Takahashi, A.; Kastan, M.B.; Griffin, D.E.; Earnshaw, W.C.; Veliuona, M.A.; et al. Modulation of Cell Death by Bcl-XL through Caspase Interaction. Proc. Natl. Acad. Sci. USA 1998, 95, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Beaumatin, F.; El Dhaybi, M.; Bobo, C.; Verdier, M.; Priault, M. Bcl-XL Deamidation and Cancer: Charting the Fame Trajectories of Legitimate Child and Hidden Siblings. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1734–1745. [Google Scholar] [CrossRef]
- Krajewska, M.; Krajewski, S. Immunohistochemical Analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 Expression in Prostate Cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar]
- Castilla, C.; Congregado, B.; Chinchón, D.; Torrubia, F.J.; Japón, M.A.; Sáez, C. Bcl-XL Is Overexpressed in Hormone-Resistant Prostate Cancer and Promotes Survival of LNCaP Cells via Interaction with Proapoptotic Bak. Endocrinology 2006, 147, 4960–4967. [Google Scholar] [CrossRef]
- Marcelli, M.; Marani, M.; Li, X.; Sturgis, L.; Haidacher, S.J.; Trial, J.; Mannucci, R.; Nicoletti, I.; Denner, L. Heterogeneous Apoptotic Responses of Prostate Cancer Cell Lines Identify an Association between Sensitivity to Staurosporine-Induced Apoptosis, Expression of Bcl-2 Family Members, and Caspase Activation. Prostate 2000, 42, 260–273. [Google Scholar] [CrossRef]
- Yancey, D.; Nelson, K.C.; Baiz, D.; Hassan, S.; Flores, A.; Pullikuth, A.; Karpova, Y.; Axanova, L.; Moore, V.; Sui, G.; et al. BAD Dephosphorylation and Decreased Expression of MCL-1 Induce Rapid Apoptosis in Prostate Cancer Cells. PLoS ONE 2013, 8, e74561. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Tang, J.; Hong, Y.; Song, J.; Terranova, P.F.; Thrasher, J.B.; Svojanovsky, S.; Wang, H.; Li, B. Androgen Receptor-Dependent Regulation of Bcl-XL Expression: Implication in Prostate Cancer Progression. Prostate 2008, 68, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Pascal, L.E.; Zhou, J.; Zhou, Y.; Wang, K.; Parwani, A.V.; Dhir, R.; Guo, P.; He, D.; Nelson, J.B.; et al. BCL-2 and BCL-XL Expression Are down-Regulated in Benign Prostate Hyperplasia Nodules and Not Affected by Finasteride and/or Celecoxib. Am. J. Clin. Exp. Urol. 2018, 6, 1–10. [Google Scholar] [PubMed]
- Lamb, L.E.; Zarif, J.C.; Miranti, C.K. The Androgen Receptor Induces Integrin A6β1 to Promote Prostate Tumor Cell Survival via NF-ΚB and Bcl-XL Independently of PI3K Signaling. Cancer Res. 2011, 71, 2739–2749. [Google Scholar] [CrossRef] [Green Version]
- Shore, G.; Warr, M. Unique Biology of Mcl-1: Therapeutic Opportunities in Cancer. Curr. Mol. Med. 2008, 8, 138–147. [Google Scholar] [CrossRef]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from Transcription to Degradation. Cell Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T. Attacking Cancer’s Achilles Heel: Antagonism of Anti-Apoptotic BCL-2 Family Members. FEBS J. 2016, 283, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhau, H.E.; Osunkoya, A.O.; Iqbal, S.; Yang, X.; Fan, S.; Chen, Z.; Wang, R.; Marshall, F.F.; Chung, L.W.; et al. Vascular Endothelial Growth Factor Regulates Myeloid Cell Leukemia-1 Expression through Neuropilin-1-Dependent Activation of c-MET Signaling in Human Prostate Cancer Cells. Mol. Cancer 2010, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Santer, F.R.; Erb, H.H.H.; Oh, S.J.; Handle, F.; Feiersinger, G.E.; Luef, B.; Bu, H.; Schäfer, G.; Ploner, C.; Egger, M.; et al. Mechanistic Rationale for MCL1 Inhibition during Androgen Deprivation Therapy. Oncotarget 2015, 6, 6105–6122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavarretta, I.T.; Neuwirt, H.; Zaki, M.H.; Steiner, H.; Hobisch, A.; Nemeth, J.A.; Culig, Z. Mcl-1 is Regulated by IL-6 and Mediates the Survival Activity of the Cytokine in a Model of Late Stage Prostate Carcinoma. In Hormonal Carcinogenesis V; Li, J.J., Li, S.A., Mohla, S., Rochefort, H., Maudelonde, T., Eds.; Advances in Experimental Medicine and, Biology; Springer: New York, NY, USA, 2008; Volume 617, pp. 547–555. ISBN 978-0-387-69078-0. [Google Scholar]
- Hassan, S.; Pullikuth, A.; Nelson, K.C.; Flores, A.; Karpova, Y.; Baiz, D.; Zhu, S.; Sui, G.; Huang, Y.; Choi, Y.A.; et al. Β2-Adrenoreceptor Signaling Increases Therapy Resistance in Prostate Cancer by Upregulating MCL1. Mol. Cancer Res. 2020, 18, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Zhang, S.; Driss, A.; Liu, Z.-R.; Kim, H.-R.C.; Wang, Y.; Ritenour, C.; Zhau, H.E.; Kucuk, O.; Chung, L.W.K.; et al. PDGF Upregulates Mcl-1 Through Activation of β-Catenin and HIF-1α-Dependent Signaling in Human Prostate Cancer Cells. PLoS ONE 2012, 7, e30764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Jonas, O.; Whitman, M.A.; Corey, E.; Balk, S.P.; Chen, S. Tyrosine Kinase Inhibitors Increase MCL1 Degradation and in Combination with BCLXL/BCL2 Inhibitors Drive Prostate Cancer Apoptosis. Clin. Cancer Res. 2018, 24, 5458–5470. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Varkaris, A.; Nouri, M.; Chen, S.; Xie, L.; Balk, S.P. MARCH5 Mediates NOXA-Dependent MCL1 Degradation Driven by Kinase Inhibitors and Integrated Stress Response Activation. eLife 2020, 9, e54954. [Google Scholar] [CrossRef]
- Dash, R.; Richards, J.E.; Su, Z.; Bhutia, S.K.; Azab, B.; Rahmani, M.; Dasmahapatra, G.; Yacoub, A.; Dent, P.; Dmitriev, I.P.; et al. Mechanism by Which Mcl-1 Regulates Cancer-Specific Apoptosis Triggered by Mda-7/IL-24, an IL-10–Related Cytokine. Cancer Res. 2010, 70, 5034–5045. [Google Scholar] [CrossRef] [Green Version]
- Pompeia, C.; Hodge, D.R.; Plass, C.; Wu, Y.-Z.; Marquez, V.E.; Kelley, J.A.; Farrar, W.L. Microarray Analysis of Epigenetic Silencing of Gene Expression in the KAS-6/1 Multiple Myeloma Cell Line. Cancer Res. 2004, 64, 3465–3473. [Google Scholar] [CrossRef] [Green Version]
- Pisani, C.; Ramella, M.; Boldorini, R.; Loi, G.; Billia, M.; Boccafoschi, F.; Volpe, A.; Krengli, M. Apoptotic and Predictive Factors by Bax, Caspases 3/9, Bcl-2, P53 and Ki-67 in Prostate Cancer after 12 Gy Single-Dose. Sci. Rep. 2020, 10, 7050. [Google Scholar] [CrossRef]
- Khor, L.Y.; Moughan, J.; Al-Saleem, T.; Hammond, E.H.; Venkatesan, V.; Rosenthal, S.A.; Ritter, M.A.; Sandler, H.M.; Hanks, G.E.; Shipley, W.U.; et al. Bcl-2 and Bax Expression Predict Prostate Cancer Outcome in Men Treated with Androgen Deprivation and Radiotherapy on Radiation Therapy Oncology Group Protocol 92-02. Clin. Cancer Res. 2007, 13, 3585–3590. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, S.; Krajewska, M.; Reed, J.C. Immunohistochemical Analysis of in Vivo Patterns of Bak Expression, a Proapoptotic Member of the Bcl-2 Protein Family. Cancer Res. 1996, 56, 2849–2855. [Google Scholar] [PubMed]
- Liu, Q.Y.; Stein, C.A. Taxol and Estramustine-Induced Modulation of Human Prostate Cancer Cell Apoptosis via Alteration in Bcl-XL and Bak Expression. Clin. Cancer Res. 1997, 3, 2039–2046. [Google Scholar] [PubMed]
- Don, M.-J.; Chang, Y.-H.; Chen, K.-K.; Ho, L.-K.; Chau, Y.-P. Induction of CDK Inhibitors (P21WAF1 and P27Kip1) and Bak in the Beta-Lapachone-Induced Apoptosis of Human Prostate Cancer Cells. Mol. Pharmacol. 2001, 59, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Lew, K.L.; Kim, Y.-A.; Zeng, Y.; Hahm, E.-R.; Dhir, R.; Singh, S.V. Diallyl Trisulfide Suppresses Growth of PC-3 Human Prostate Cancer Xenograft In Vivo in Association with Bax and Bak Induction. Clin. Cancer Res. 2006, 12, 6836–6843. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.L.; Storey, A. BMX Negatively Regulates BAK Function, Thereby Increasing Apoptotic Resistance to Chemotherapeutic Drugs. Cancer Res. 2015, 75, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cai, C.; Sowalsky, A.G.; Ye, H.; Ma, F.; Yuan, X.; Simon, N.I.; Gray, N.S.; Balk, S.P. BMX-Mediated Regulation of Multiple Tyrosine Kinases Contributes to Castration Resistance in Prostate Cancer. Cancer Res. 2018, 78, 5203–5215. [Google Scholar] [CrossRef] [Green Version]
- Lomonosova, E.; Chinnadurai, G. BH3-Only Proteins in Apoptosis and beyond: An Overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, N.L.; Pandey, V.; Zhu, T.; Ma, L.; Basappa; Lobie, P.E. Bad Phosphorylation as a Target of Inhibition in Oncology. Cancer Lett. 2018, 415, 177–186. [Google Scholar] [CrossRef]
- Danial, N.N. BAD: Undertaker by Night, Candyman by Day. Oncogene 2008, 27, S53–S70. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Royuela, M.; Arenas, M.I.; Bethencourt, F.R.; Sanchez-Chapado, M.; Fraile, B.; Paniagua, R. Immunoexpressions of P21, Rb, Mcl-1 and Bad Gene Products in Normal, Hyperplastic and Carcinomatous Human Prostates. Eur. Cytokine Netw. 2001, 12, 654–663. [Google Scholar] [PubMed]
- Teo, K.; Gemmell, L.; Mukherjee, R.; Traynor, P.; Edwards, J. Bad Expression Influences Time to Androgen Escape in Prostate Cancer. BJU Int. 2007, 100, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Chao, O.S.P.; Clément, M.-V. Epidermal Growth Factor and Serum Activate Distinct Pathways to Inhibit the BH3 Only Protein BAD in Prostate Carcinoma LNCaP Cells. Oncogene 2006, 25, 4458–4469. [Google Scholar] [CrossRef]
- Meshki, J.; Caino, M.C.; von Burstin, V.A.; Griner, E.; Kazanietz, M.G. Regulation of Prostate Cancer Cell Survival by Protein Kinase Cϵ Involves Bad Phosphorylation and Modulation of the TNFα/JNK Pathway. J. Biol. Chem. 2010, 285, 26033–26040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumori, T.; Oka, N.; Takenaka, Y.; Nangia-Makker, P.; Elsamman, E.; Kasai, T.; Shono, M.; Kanayama, H.; Ellerhorst, J.; Lotan, R.; et al. Galectin-3 Regulates Mitochondrial Stability and Antiapoptotic Function in Response to Anticancer Drug in Prostate Cancer. Cancer Res. 2006, 66, 3114–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Activation and Cross-Talk between Akt, NF-ΚB, and Unfolded Protein Response Signaling in 1-LN Prostate Cancer Cells Consequent to Ligation of Cell Surface-Associated GRP78. J. Biol. Chem. 2006, 281, 13694–13707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, K.S.R.; Smith, A.J.; Karpova, Y.; Datta, S.R.; Kulik, G. Diverse Antiapoptotic Signaling Pathways Activated by Vasoactive Intestinal Polypeptide, Epidermal Growth Factor, and Phosphatidylinositol 3-Kinase in Prostate Cancer Cells Converge on BAD. J. Biol. Chem. 2006, 281, 20891–20901. [Google Scholar] [CrossRef] [Green Version]
- Sastry, K.S.R.; Karpova, Y.; Kulik, G. Epidermal Growth Factor Protects Prostate Cancer Cells from Apoptosis by Inducing BAD Phosphorylation via Redundant Signaling Pathways. J. Biol. Chem. 2006, 281, 27367–27377. [Google Scholar] [CrossRef] [Green Version]
- Sastry, K.S.R.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine Protects Cancer Cells from Apoptosis via Activation of CAMP-Dependent Protein Kinase and BAD Phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berquin, I.M.; Min, Y.; Wu, R.; Wu, J.; Perry, D.; Cline, J.M.; Thomas, M.J.; Thornburg, T.; Kulik, G.; Smith, A.; et al. Modulation of Prostate Cancer Genetic Risk by Omega-3 and Omega-6 Fatty Acids. J. Clin. Invest. 2007, 117, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Ammar, H.; Closset, J.L. Clusterin Activates Survival through the Phosphatidylinositol 3-Kinase/Akt Pathway. J. Biol. Chem. 2008, 283, 12851–12861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumenthaler, S.M.; Ng, P.Y.B.; Hodge, A.; Bearss, D.; Berk, G.; Kanekal, S.; Redkar, S.; Taverna, P.; Agus, D.B.; Jain, A. Pharmacologic Inhibition of Pim Kinases Alters Prostate Cancer Cell Growth and Resensitizes Chemoresistant Cells to Taxanes. Mol. Cancer Ther. 2009, 8, 2882–2893. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Siedow, M.; Saia, G.; Chakravarti, A. Inhibition of P21-Activated Kinase 6 (PAK6) Increases Radiosensitivity of Prostate Cancer Cells: PAK6 and Prostate Cancer. Prostate 2010, 70, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Zoubeidi, A.; Zardan, A.; Wiedmann, R.M.; Locke, J.; Beraldi, E.; Fazli, L.; Gleave, M.E. Hsp27 Promotes Insulin-Like Growth Factor-I Survival Signaling in Prostate Cancer via P90Rsk-Dependent Phosphorylation and Inactivation of BAD. Cancer Res. 2010, 70, 2307–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, T.; Koreckij, T.; Corey, E. Targeted Therapy for Advanced Prostate Cancer: Inhibition of the PI3K/Akt/MTOR Pathway. Curr. Cancer Drug Targets 2009, 9, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Demeter, M.R.; Ruan, H.; Comb, M.J. BAD Ser-155 Phosphorylation Regulates BAD/Bcl-XL Interaction and Cell Survival. J. Biol. Chem. 2000, 275, 25865–25869. [Google Scholar] [CrossRef] [Green Version]
- Dramsi, S.; Scheid, M.P.; Maiti, A.; Hojabrpour, P.; Chen, X.; Schubert, K.; Goodlett, D.R.; Aebersold, R.; Duronio, V. Identification of a Novel Phosphorylation Site, Ser-170, as a Regulator of Bad Pro-apoptotic Activity. J. Biol. Chem. 2002, 277, 6399–6405. [Google Scholar] [CrossRef] [Green Version]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-W.; Chandra, D.; Tang, S.-H.; Chopra, D.; Tang, D.G. Identification and Characterization of Bimgamma, a Novel Proapoptotic BH3-Only Splice Variant of Bim. Cancer Res. 2002, 62, 2976–2981. [Google Scholar] [PubMed]
- Tan, T.-T.; Degenhardt, K.; Nelson, D.A.; Beaudoin, B.; Nieves-Neira, W.; Bouillet, P.; Villunger, A.; Adams, J.M.; White, E. Key Roles of BIM-Driven Apoptosis in Epithelial Tumors and Rational Chemotherapy. Cancer Cell 2005, 7, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Aira, L.E.; Villa, E.; Colosetti, P.; Gamas, P.; Signetti, L.; Obba, S.; Proics, E.; Gautier, F.; Bailly-Maitre, B.; Jacquel, A.; et al. The Oncogenic Tyrosine Kinase Lyn Impairs the Pro-Apoptotic Function of Bim. Oncogene 2018, 37, 2122–2136. [Google Scholar] [CrossRef]
- Gogada, R.; Yadav, N.; Liu, J.; Tang, S.; Zhang, D.; Schneider, A.; Seshadri, A.; Sun, L.; Aldaz, C.M.; Tang, D.G.; et al. Bim, a Proapoptotic Protein, Up-Regulated via Transcription Factor E2F1-Dependent Mechanism, Functions as a Prosurvival Molecule in Cancer. J. Biol. Chem. 2013, 288, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-C.; Lin, R.-W.; Huang, S.-B.; Huang, S.-Y.; Chen, W.-J.; Wang, S.; Hong, Y.-R.; Wang, C. Bim Directly Antagonizes Bcl-Xl in Doxorubicin-Induced Prostate Cancer Cell Apoptosis Independently of P53. Cell Cycle 2016, 15, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Lin, Y.; Cui, P.; Li, H.; Zhang, L.; Sun, Z.; Huang, S.; Li, S.; Huang, S.; Zhao, Q.; et al. Cdc20/P55 Mediates the Resistance to Docetaxel in Castration-Resistant Prostate Cancer in a Bim-Dependent Manner. Cancer Chemother. Pharmacol. 2018, 81, 999–1006. [Google Scholar] [CrossRef]
- Kinkade, C.W.; Castillo-Martin, M.; Puzio-Kuter, A.; Yan, J.; Foster, T.H.; Gao, H.; Sun, Y.; Ouyang, X.; Gerald, W.L.; Cordon-Cardo, C.; et al. Targeting AKT/MTOR and ERK MAPK Signaling Inhibits Hormone-Refractory Prostate Cancer in a Preclinical Mouse Model. J. Clin. Invest. 2008, 3051–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, L. PUMA, a Potent Killer with or without P53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Kuwahara, M.; Suzuki, A.; Hirahara, K.; Shinnaksu, R.; Hosokawa, H.; Hasegawa, A.; Motohashi, S.; Iwama, A.; Nakayama, T. Bmi1 Regulates Memory CD4 T Cell Survival via Repression of the Noxa Gene. J. Exp. Med. 2008, 205, 1109–1120. [Google Scholar] [CrossRef]
- Albert, M.-C.; Brinkmann, K.; Kashkar, H. Noxa and Cancer Therapy: Tuning up the Mitochondrial Death Machinery in Response to Chemotherapy. Mol. Cell. Oncol. 2014, 1, e29906. [Google Scholar] [CrossRef] [Green Version]
- Diallo, J.-S.; Aldejmah, A.; Mouhim, A.F.; Péant, B.; Fahmy, M.A.; Koumakpayi, I.H.; Sircar, K.; Bégin, L.R.; Mes-Masson, A.-M.; Saad, F. NOXA and PUMA Expression Add to Clinical Markers in Predicting Biochemical Recurrence of Prostate Cancer Patients in a Survival Tree Model. Clin. Cancer Res. 2007, 13, 7044–7052. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Ström, A.; Gustafsson, J.-Å. Estrogen Receptor β Upregulates FOXO3a and Causes Induction of Apoptosis through PUMA in Prostate Cancer. Oncogene 2014, 33, 4213–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Tang, W.; Dai, Y.; Wu, X.; Liu, M.; Ji, Q.; Ji, M.; Pienta, K.; Lawrence, T.; Xu, L. Natural BH3 Mimetic (-)-Gossypol Chemosensitizes Human Prostate Cancer via Bcl-XL Inhibition Accompanied by Increase of Puma and Noxa. Mol. Cancer Ther. 2008, 7, 2192–2202. [Google Scholar] [CrossRef] [Green Version]
- Pang, C.-Y.; Chiu, S.-C.; Harn, H.-J.; Zhai, W.-J.; Lin, S.-Z.; Yang, H.-H. Proteomic-Based Identification of Multiple Pathways Underlying n-Butylidenephthalide-Induced Apoptosis in LNCaP Human Prostate Cancer Cells. Food Chem. Toxicol. 2013, 59, 281–288. [Google Scholar] [CrossRef]
- Nikrad, M.; Johnson, T.; Puthalalath, H.; Coultas, L.; Adams, J.; Kraft, A.S. The Proteasome Inhibitor Bortezomib Sensitizes Cells to Killing by Death Receptor Ligand TRAIL via BH3-Only Proteins Bik and Bim. Mol. Cancer Ther. 2005, 4, 443–449. [Google Scholar] [CrossRef]
- Hockings, C.; Anwari, K.; Ninnis, R.L.; Brouwer, J.; O’Hely, M.; Evangelista, M.; Hinds, M.G.; Czabotar, P.E.; Lee, E.F.; Fairlie, W.D.; et al. Bid Chimeras Indicate That Most BH3-Only Proteins Can Directly Activate Bak and Bax, and Show No Preference for Bak versus Bax. Cell Death Dis. 2015, 6, e1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; O’Neill, K.L.; Huang, K. The Third Model of Bax/Bak Activation: A Bcl-2 Family Feud Finally Resolved? F1000Res 2020, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Sax, J.K.; Fei, P.; Murphy, M.E.; Bernhard, E.; Korsmeyer, S.J.; El-Deiry, W.S. BID Regulation by P53 Contributes to Chemosensitivity. Nat. Cell Biol. 2002, 4, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-Mediated Down-Regulation of Bid and Bax in Tumors Occurs via Hypoxia-Inducible Factor 1-Dependent and -Independent Mechanisms and Contributes to Drug Resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewska, M.; Zapata, J.M.; Meinhold-Heerlein, I.; Hedayat, H.; Monks, A.; Bettendorf, H.; Shabaik, A.; Bubendorf, L.; Kallioniemi, O.-P.; Kim, H.; et al. Expression of Bcl-2 Family Member Bid in Normal and Malignant Tissues. Neoplasia 2002, 4, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Nesterov, A.; Lu, X.; Johnson, M.; Miller, G.J.; Ivashchenko, Y.; Kraft, A.S. Elevated Akt Activity Protects the Prostate Cancer Cell Line LNCaP from TRAIL-Induced Apoptosis. J. Biol. Chem. 2001, 276, 10767–10774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmanapalli, R.; Perkins, C.L.; Orlando, M.; O’Bryan, E.; Nguyen, D.; Bhalla, K.N. Pretreatment with Paclitaxel Enhances Apo-2 Ligand/Tumor Necrosis Factor- Related Apoptosis-Inducing Ligand-Induced Apoptosis of Prostate Cancer Cells by Inducing Death Receptors 4 and 5 Protein Levels. Cancer Res. 2001, 61, 759–763. [Google Scholar]
- Kulik, G.; Carson, J.P.; Vomastek, T.; Overman, K.; Gooch, B.D.; Srinivasula, S.; Alnemri, E.; Nunez, G.; Weber, M.J. Tumor Necrosis Factor Alpha Induces BID Cleavage and Bypasses Antiapoptotic Signals in Prostate Cancer LNCaP Cells. Cancer Res. 2001, 61, 2713–2719. [Google Scholar] [PubMed]
- Rokhlin, O.W.; Guseva, N.; Tagiyev, A.; Knudson, C.M.; Cohen, M.B. Bcl-2 Oncoprotein Protects the Human Prostatic Carcinoma Cell Line PC3 from TRAIL-Mediated Apoptosis. Oncogene 2001, 20, 2836–2843. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Thakkar, H.; Tyan, F.; Gim, S.; Robinson, H.; Lee, C.; Pandey, S.K.; Nwokorie, C.; Onwudiwe, N.; Srivastava, R.K. Constitutively Active Akt Is an Important Regulator of TRAIL Sensitivity in Prostate Cancer. Oncogene 2001, 20, 6073–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Pellegrini, K.; Ouellet, V.; Giuste, F.; Ramalingam, S.; Watanabe, K.; Adam-Granger, E.; Fossouo, L.; You, S.; Freeman, M.; et al. Identification of the Transcription Factor Relationships Associated with Androgen Deprivation Therapy Response and Metastatic Progression in Prostate Cancer. Cancers 2018, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, Y.; Feng, W.; Wang, X.; Yang, J.Y.; Zhao, Y.; Wang, Y.; Liu, Y. Transcription Factor and MicroRNA Regulation in Androgen-Dependent and -Independent Prostate Cancer Cells. BMC Genom. 2008, 9, S22. [Google Scholar] [CrossRef] [Green Version]
- Volante, M.; Tota, D.; Giorcelli, J.; Bollito, E.; Napoli, F.; Vatrano, S.; Buttigliero, C.; Molinaro, L.; Gontero, P.; Porpiglia, F.; et al. Androgen Deprivation Modulates Gene Expression Profile along Prostate Cancer Progression. Hum. Pathol. 2016, 56, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Henderson, V.; Smith, B.; Burton, L.J.; Randle, D.; Morris, M.; Odero-Marah, V.A. Snail Promotes Cell Migration through PI3K/AKT-Dependent Rac1 Activation as Well as PI3K/AKT-Independent Pathways during Prostate Cancer Progression. Cell Adhes. Migr. 2015, 9, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Porter, G.W.; Khuri, F.R.; Fu, H. Dynamic 14-3-3/Client Protein Interactions Integrate Survival and Apoptotic Pathways. Semin. Cancer Biol. 2006, 16, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Peso, L.D. Interleukin-3-Induced Phosphorylation of BAD Through the Protein Kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef]
- Qi, X.-J.; Wildey, G.M.; Howe, P.H. Evidence That Ser87 of BimEL Is Phosphorylated by Akt and Regulates BimEL Apoptotic Function. J. Biol. Chem. 2006, 281, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslan, J.E.; You, H.; Williamson, D.M.; Endig, J.; Youker, R.T.; Thomas, L.; Shu, H.; Du, Y.; Milewski, R.L.; Brush, M.H.; et al. Akt and 14-3-3 Control a PACS-2 Homeostatic Switch That Integrates Membrane Traffic with TRAIL-Induced Apoptosis. Mol. Cell 2009, 34, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.; Pellegrini, M.; Tsuchihara, K.; Yamamoto, K.; Hacker, G.; Erlacher, M.; Villunger, A.; Mak, T.W. FOXO3a-Dependent Regulation of Puma in Response to Cytokine/Growth Factor Withdrawal. J. Exp. Med. 2006, 203, 1657–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S.; Fujita, N.; Kitahara, O.; Nakamura, Y.; Tsuruo, T. Involvement of FKHR-Dependent TRADD Expression in Chemotherapeutic Drug-Induced Apoptosis. Mol. Cell. Biol. 2002, 22, 8695–8708. [Google Scholar] [CrossRef] [Green Version]
- Dijkers, P.F.; Medema, R.H.; Lammers, J.-W.J.; Koenderman, L.; Coffer, P.J. Expression of the Pro-Apoptotic Bcl-2 Family Member Bim Is Regulated by the Forkhead Transcription Factor FKHR-L1. Curr. Biol. 2000, 10, 1201–1204. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-M.; Chao, J.-R.; Chen, W.; Kuo, M.-L.; Yen, J.J.-Y.; Yang-Yen, H.-F. The Antiapoptotic Gene Mcl-1 Is Up-Regulated by the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway through a Transcription Factor Complex Containing CREB. Mol. Cell. Biol. 1999, 19, 6195–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, M.-L.; Chuang, S.-E.; Lin, M.-T.; Yang, S.-Y. The Involvement of PI 3-K/Akt-Dependent up-Regulation of Mcl-1 in the Prevention of Apoptosis of Hep3B Cells by Interleukin-6. Oncogene 2001, 20, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Crawford, L.; Leeds, J.C.; Nussbaum, D.P.; Shankar, P.S.; Soderquist, R.S.; Stein, E.M.; Tingley, J.P.; et al. PIK3CA Mutations Enable Targeting of a Breast Tumor Dependency through MTOR-Mediated MCL-1 Translation. Sci. Transl. Med. 2016, 8, 369ra175. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen Synthase Kinase-3 Regulates Mitochondrial Outer Membrane Permeabilization and Apoptosis by Destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef]
- Wozney, J.L.; Antonarakis, E.S. Growth Factor and Signaling Pathways and Their Relevance to Prostate Cancer Therapeutics. Cancer Metastasis Rev. 2014, 33, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M.; Abate-Shen, C. Pten Inactivation and the Emergence of Androgen-Independent Prostate Cancer. Cancer Res. 2007, 67, 6535–6538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Berriguete, G.; Fraile, B.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Royuela, M. MAP Kinases and Prostate Cancer. J. Signal Transduct. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 Activation Controls Cell Proliferation and Cell Death: Is Subcellular Localization the Answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzmaier, F.J.; Ramos, J.W. RSK Isoforms in Cancer Cell Invasion and Metastasis. Cancer Res. 2013, 73, 6099–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Ptenloss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK Signaling Is a Therapeutic Target in Metastatic Castration Resistant Prostate Cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F.; Weber, M.J. Activation of Mitogen-Activated Protein Kinase Associated with Prostate Cancer Progression. Cancer Res. 1999, 59, 279–284. [Google Scholar] [PubMed]
- Mukherjee, R.; McGuinness, D.H.; McCall, P.; Underwood, M.A.; Seywright, M.; Orange, C.; Edwards, J. Upregulation of MAPK Pathway Is Associated with Survival in Castrate-Resistant Prostate Cancer. Br. J. Cancer 2011, 104, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ouyang, X.; Banach-Petrosky, W.A.; Gerald, W.L.; Shen, M.M.; Abate-Shen, C. Combinatorial Activities of Akt and B-Raf/Erk Signaling in a Mouse Model of Androgen-Independent Prostate Cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 14477–14482. [Google Scholar] [CrossRef] [Green Version]
- Labanca, E.; Vazquez, E.S.; Corn, P.G.; Roberts, J.M.; Wang, F.; Logothetis, C.J.; Navone, N.M. Fibroblast Growth Factors Signaling in Bone Metastasis. Endocr. Relat. Cancer 2020, 27, R255–R265. [Google Scholar] [CrossRef]
- Sastry, K.S.; Chouchane, A.I.; Wang, E.; Kulik, G.; Marincola, F.M.; Chouchane, L. Cytoprotective Effect of Neuropeptides on Cancer Stem Cells: Vasoactive Intestinal Peptide-Induced Antiapoptotic Signaling. Cell Death Dis. 2017, 8, e2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, W. Beta-Adrenergic Signaling on Neuroendocrine Differentiation, Angiogenesis, and Metastasis in Prostate Cancer Progression. Asian J. Androl. 2019, 21, 253. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Taskén, K.A. β-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, I. Mechanisms Involved in VPAC Receptors Activation and Regulation: Lessons from Pharmacological and Mutagenesis Studies. Front. Endocrinol. 2012, 3, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, B.; Carmena, M.; Sánchez-Chapado, M.; Ruíz-Villaespesa, A.; Bajo, A.; Fernández-Martínez, A.; Varga, J.; Schally, A.; Prieto, J. Expression of Vasoactive Intestinal Peptide and Functional VIP Receptors in Human Prostate Cancer: Antagonistic Action of a Growth-Hormone-Releasing Hormone Analog. Int. J. Oncol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wolff, D.W.; Lin, M.-F.; Tu, Y. Vasoactive Intestinal Peptide Transactivates the Androgen Receptor through a Protein Kinase A-Dependent Extracellular Signal-Regulated Kinase Pathway in Prostate Cancer LNCaP Cells. Mol. Pharmacol. 2007, 72, 73–85. [Google Scholar] [CrossRef]
- Lemeshow, S.; Sørensen, H.T.; Phillips, G.; Yang, E.V.; Antonsen, S.; Riis, A.H.; Lesinski, G.B.; Jackson, R.; Glaser, R. β-Blockers and Survival among Danish Patients with Malignant Melanoma: A Population-Based Cohort Study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2273–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, V.D.; Grazzini, M.; Gandini, S.; Benemei, S.; Lotti, T.; Marchionni, N.; Geppetti, P. Treatment with β-Blockers and Reduced Disease Progression in Patients with Thick Melanoma. Arch. Intern. Med. 2011, 171, 3. [Google Scholar] [CrossRef]
- Baek, M.-H.; Kim, D.-Y.; Kim, S.O.; Kim, Y.-J.; Park, Y.-H. Impact of Beta Blockers on Survival Outcomes in Ovarian Cancer: A Nationwide Population-Based Cohort Study. J. Gynecol. Oncol. 2018, 29, e82. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of Host Bone Marrow Stromal Cells by Sympathetic Nerves Promotes Breast Cancer Bone Metastasis in Mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, D.; Lang, K.; Niggemann, B.; Drell, T.L.; Masur, K.; Zaenker, K.S.; Entschladen, F. The Norepinephrine-Driven Metastasis Development of PC-3 Human Prostate Cancer Cells in BALB/c Nude Mice Is Inhibited by β-Blockers. Int. J. Cancer 2006, 118, 2744–2749. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A. Association Between Use of β-Blockers and Prostate Cancer–Specific Survival: A Cohort Study of 3561 Prostate Cancer Patients with High-Risk or Metastatic Disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A.; Håheim, L.L. Use of β-Blockers Is Associated with Prostate Cancer-Specific Survival in Prostate Cancer Patients on Androgen Deprivation Therapy: β-Blocker Use and Prostate Cancer Survival. Prostate 2013, 73, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohée, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol Sensitizes Prostate Cancer Cells to Glucose Metabolism Inhibition and Prevents Cancer Progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [PubMed]
- Asdaq, S.M.B.; Inamdar, M.N. Pharmacodynamic and Pharmacokinetic Interactions of Propranolol with Garlic (Allium Sativum) in Rats. Evid. Based Complement. Altern. Med. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II Trial of the PI3 Kinase Inhibitor Buparlisib (BKM-120) with or without Enzalutamide in Men with Metastatic Castration Resistant Prostate Cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef]
- Hotte, S.J.; Chi, K.N.; Joshua, A.M.; Tu, D.; Macfarlane, R.J.; Gregg, R.W.; Ruether, J.D.; Basappa, N.S.; Finch, D.; Salim, M.; et al. A Phase II Study of PX-866 in Patients with Recurrent or Metastatic Castration-Resistant Prostate Cancer: Canadian Cancer Trials Group Study IND205. Clin. Genitourin. Cancer 2019, 17, 201–208.e1. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A Phase II Study of the Dual MTOR Inhibitor MLN0128 in Patients with Metastatic Castration Resistant Prostate Cancer. Invest. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Shen, T.; Halabi, S.; Kemeny, G.; Bitting, R.L.; Kartcheske, P.; Embree, E.; Morris, K.; Winters, C.; Jaffe, T.; et al. A Phase II Trial of Temsirolimus in Men with Castration-Resistant Metastatic Prostate Cancer. Clin. Genitourin. Cancer 2013, 11, 397–406. [Google Scholar] [CrossRef]
- Emmenegger, U.; Booth, C.M.; Berry, S.; Sridhar, S.S.; Winquist, E.; Bandali, N.; Chow, A.; Lee, C.; Xu, P.; Man, S.; et al. Temsirolimus Maintenance Therapy After Docetaxel Induction in Castration-Resistant Prostate Cancer. Oncologist 2015, 20, 1351–1352. [Google Scholar] [CrossRef] [Green Version]
- Kruczek, K.; Ratterman, M.; Tolzien, K.; Sulo, S.; Lestingi, T.M.; Nabhan, C. A Phase II Study Evaluating the Toxicity and Efficacy of Single-Agent Temsirolimus in Chemotherapy-Naïve Castration-Resistant Prostate Cancer. Br. J. Cancer 2013, 109, 1711–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meulenbeld, H.J.; de Bono, J.S.; Tagawa, S.T.; Whang, Y.E.; Li, X.; Heath, K.H.; Zandvliet, A.S.; Ebbinghaus, S.W.; Hudes, G.R.; de Wit, R. Tolerability, Safety and Pharmacokinetics of Ridaforolimus in Combination with Bicalutamide in Patients with Asymptomatic, Metastatic Castration-Resistant Prostate Cancer (CRPC). Cancer Chemother. Pharmacol. 2013, 72, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.; Shevrin, D.; Stein, M.; Heilbrun, L.; Land, S.; Stark, K.; Li, J.; Dickow, B.; Heath, E.; Smith, D.; et al. Phase II Trial of Carboplatin, Everolimus, and Prednisone in Metastatic Castration-Resistant Prostate Cancer Pretreated with Docetaxel Chemotherapy: A Prostate Cancer Clinical Trial Consortium Study. Urology 2015, 86, 1206–1211. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bärtschi, D.; Dröge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Phase 2 Trial of Single-Agent Everolimus in Chemotherapy-Naive Patients with Castration-Resistant Prostate Cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Werner, L.; Courtney, K.D.; Buckle, G.; Oh, W.K.; Bubley, G.J.; Hayes, J.H.; Weckstein, D.; Elfiky, A.; Sims, D.M.; et al. Phase II Trial of RAD001 and Bicalutamide for Castration-Resistant Prostate Cancer: Phase II Trial of RAD001 and Bicalutamide. BJU Int. 2012, 110, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.; Ghosh, P.M.; de Vere White, R.; Evans, C.P.; Dall’Era, M.A.; Yap, S.A.; Li, Y.; Beckett, L.A.; Lara, P.N.; Pan, C.-X. A Phase 2 Clinical Trial of Everolimus plus Bicalutamide for Castration-Resistant Prostate Cancer: Everolimus and Bicalutamide for CRPC. Cancer 2016, 122, 1897–1904. [Google Scholar] [CrossRef] [Green Version]
- Rathkopf, D.E.; Larson, S.M.; Anand, A.; Morris, M.J.; Slovin, S.F.; Shaffer, D.R.; Heller, G.; Carver, B.; Rosen, N.; Scher, H.I. Everolimus Combined with Gefitinib in Patients with Metastatic Castration-Resistant Prostate Cancer: Phase 1/2 Results and Signaling Pathway Implications: Everolimus and Gefitinib in CRPC. Cancer 2015, 121, 3853–3861. [Google Scholar] [CrossRef]
- Amato, R.J.; Wilding, G.; Bubley, G.; Loewy, J.; Haluska, F.; Gross, M.E. Safety and Preliminary Efficacy Analysis of the MTOR Inhibitor Ridaforolimus in Patients with Taxane-Treated, Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2012, 10, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Rathi, N.; Maughan, B.L.; Agarwal, N.; Swami, U. Mini-Review: Cabozantinib in the Treatment of Advanced Renal Cell Carcinoma and Hepatocellular Carcinoma. Cancer Manag. Res. 2020, 12, 3741–3749. [Google Scholar] [CrossRef]
- Smith, D.C.; Smith, M.R.; Sweeney, C.; Elfiky, A.A.; Logothetis, C.; Corn, P.G.; Vogelzang, N.J.; Small, E.J.; Harzstark, A.L.; Gordon, M.S.; et al. Cabozantinib in Patients with Advanced Prostate Cancer: Results of a Phase II Randomized Discontinuation Trial. J. Clin. Oncol. 2013, 31, 412–419. [Google Scholar] [CrossRef]
- Schöffski, P.; Gordon, M.; Smith, D.C.; Kurzrock, R.; Daud, A.; Vogelzang, N.J.; Lee, Y.; Scheffold, C.; Shapiro, G.I. Phase II Randomised Discontinuation Trial of Cabozantinib in Patients with Advanced Solid Tumours. Eur. J. Cancer 2017, 86, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Sweeney, C.J.; Corn, P.G.; Rathkopf, D.E.; Smith, D.C.; Hussain, M.; George, D.J.; Higano, C.S.; Harzstark, A.L.; Sartor, A.O.; et al. Cabozantinib in Chemotherapy-Pretreated Metastatic Castration-Resistant Prostate Cancer: Results of a Phase II Nonrandomized Expansion Study. J. Clin. Oncol. 2014, 32, 3391–3399. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef]
- Basch, E.M.; Scholz, M.; de Bono, J.S.; Vogelzang, N.; de Souza, P.; Marx, G.; Vaishampayan, U.; George, S.; Schwarz, J.K.; Antonarakis, E.S.; et al. Cabozantinib Versus Mitoxantrone-Prednisone in Symptomatic Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase 3 Trial with a Primary Pain Endpoint. Eur. Urol. 2019, 75, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Molife, L.R.; Omlin, A.; Jones, R.J.; Karavasilis, V.; Bloomfield, D.; Lumsden, G.; Fong, P.C.; Olmos, D.; O’Sullivan, J.M.; Pedley, I.; et al. Randomized Phase II Trial of Nintedanib, Afatinib and Sequential Combination in Castration-Resistant Prostate Cancer. Future Oncol. 2014, 10, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Dror Michaelson, M.; Regan, M.M.; Oh, W.K.; Kaufman, D.S.; Olivier, K.; Michaelson, S.Z.; Spicer, B.; Gurski, C.; Kantoff, P.W.; Smith, M.R. Phase II Study of Sunitinib in Men with Advanced Prostate Cancer. Ann. Oncol. 2009, 20, 913–920. [Google Scholar] [CrossRef]
- Canil, C.M.; Moore, M.J.; Winquist, E.; Baetz, T.; Pollak, M.; Chi, K.N.; Berry, S.; Ernst, D.S.; Douglas, L.; Brundage, M.; et al. Randomized Phase II Study of Two Doses of Gefitinib in Hormone-Refractory Prostate Cancer: A Trial of the National Cancer Institute of Canada-Clinical Trials Group. J. Clin. Oncol. 2005, 23, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Braud, F.D.; Sandri, M.T.; Renne, G.; Zorzino, L.; Scardino, E.; Rocco, B.; Spitaleri, G.; Pas, T.D.; Noberasco, C.; et al. Gefitinib Combined with Endocrine Manipulation in Patients with Hormone-Refractory Prostate Cancer: Quality of Life and Surrogate Markers of Activity. Anticancer Drugs 2007, 18, 949–954. [Google Scholar] [CrossRef]
- Curigliano, G.; Pelosi, G.; De Pas, T.; Renne, G.; De Cobelli, O.; Manzotti, M.; Spitaleri, G.; de Braud, F. Absence of Epidermal Growth Factor Receptor Gene Mutations in Patients with Hormone Refractory Prostate Cancer Not Responding to Gefitinib. Prostate 2007, 67, 603–604. [Google Scholar] [CrossRef]
- Salzberg, M.; Rochlitz, C.; Morant, R.; Thalmann, G.; Pedrazzini, A.; Roggero, E.; Schönenberger, A.; Knuth, A.; Borner, M. An Open-Label, Noncomparative Phase II Trial to Evaluate the Efficacy and Safety of Docetaxel in Combination with Gefitinib in Patients with Hormone-Refractory Metastatic Prostate Cancer. Oncol. Res. Treat. 2007, 30, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Fontana, J.; Tannir, N.; DiPaola, R.S.; Wilding, G.; Rubin, M.; Iacona, R.B.; Kabbinavar, F.F. A Phase II Trial of Gefitinib in Patients with Non-Metastatic Hormone-Refractory Prostate Cancer. BJU Int. 2007, 100, 765–769. [Google Scholar] [CrossRef]
- Curigliano, G.; Spitaleri, G.; Cobelli, O.D.; Scardino, E.; Sbanotto, A.; Braud, F.D. Health-Related Quality of Life in Patients with Hormone Refractory Prostate Cancer Receiving Gefitinib. Urol. Int. 2009, 82, 196–202. [Google Scholar] [CrossRef]
- Boccardo, F.; Rubagotti, A.; Conti, G.; Battaglia, M.; Cruciani, G.; Manganelli, A.; Ricci, S.; Lapini, A. Prednisone plus Gefitinib versus Prednisone plus Placebo in the Treatment of Hormone-Refractory Prostate Cancer: A Randomized Phase II Trial. Oncology 2008, 74, 223–228. [Google Scholar] [CrossRef]
- Whang, Y.E.; Armstrong, A.J.; Rathmell, W.K.; Godley, P.A.; Kim, W.Y.; Pruthi, R.S.; Wallen, E.M.; Crane, J.M.; Moore, D.T.; Grigson, G.; et al. A Phase II Study of Lapatinib, a Dual EGFR and HER-2 Tyrosine Kinase Inhibitor, in Patients with Castration-Resistant Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Higano, C.; Pantuck, A.; Castellanos, O.; Green, E.; Nguyen, K.; Agus, D.B. A Phase II Trial of Docetaxel and Erlotinib as First-Line Therapy for Elderly Patients with Androgen-Independent Prostate Cancer. BMC Cancer 2007, 7, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, C.; Lestingi, T.M.; Galvez, A.; Tolzien, K.; Kelby, S.K.; Tsarwhas, D.; Newman, S.; Bitran, J.D. Erlotinib Has Moderate Single-Agent Activity in Chemotherapy-Naïve Castration-Resistant Prostate Cancer: Final Results of a Phase II Trial. Urology 2009, 74, 665–671. [Google Scholar] [CrossRef]
- Gravis, G.; Bladou, F.; Salem, N.; Gonçalves, A.; Esterni, B.; Walz, J.; Bagattini, S.; Marcy, M.; Brunelle, S.; Viens, P. Results from a Monocentric Phase II Trial of Erlotinib in Patients with Metastatic Prostate Cancer. Ann. Oncol. 2008, 19, 1624–1628. [Google Scholar] [CrossRef]
- Azad, A.A.; Beardsley, E.K.; Hotte, S.J.; Ellard, S.L.; Klotz, L.; Chin, J.; Kollmannsberger, C.; Mukherjee, S.D.; Chi, K.N. A Randomized Phase II Efficacy and Safety Study of Vandetanib (ZD6474) in Combination with Bicalutamide versus Bicalutamide Alone in Patients with Chemotherapy Naïve Castration-Resistant Prostate Cancer. Invest. New Drugs 2014, 32, 746–752. [Google Scholar] [CrossRef]
- Horti, J.; Widmark, A.; Stenzl, A.; Federico, M.H.; Abratt, R.P.; Sanders, N.; Pover, G.M.; Bodrogi, I. A Randomized, Double-Blind, Placebo-Controlled Phase II Study of Vandetanib Plus Docetaxel/Prednisolone in Patients with Hormone-Refractory Prostate Cancer. Cancer Biother. Radiopharm. 2009, 24, 175–180. [Google Scholar] [CrossRef]
- Cathomas, R.; Rothermundt, C.; Klingbiel, D.; Bubendorf, L.; Jaggi, R.; Betticher, D.C.; Brauchli, P.; Cotting, D.; Droege, C.; Winterhalder, R.; et al. Efficacy of Cetuximab in Metastatic Castration-Resistant Prostate Cancer Might Depend on EGFR and PTEN Expression: Results from a Phase II Trial (SAKK 08/07). Clin. Cancer Res. 2012, 18, 6049–6057. [Google Scholar] [CrossRef] [Green Version]
- Slovin, S.F.; Kelly, W.K.; Wilton, A.; Kattan, M.; Myskowski, P.; Mendelsohn, J.; Scher, H.I. Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab Plus Doxorubicin in the Treatment of Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2009, 7, E77–E82. [Google Scholar] [CrossRef]
- Fleming, M.T.; Sonpavde, G.; Kolodziej, M.; Awasthi, S.; Hutson, T.E.; Martincic, D.; Rastogi, A.; Rousey, S.R.; Weinstein, R.E.; Galsky, M.D.; et al. Association of Rash with Outcomes in a Randomized Phase II Trial Evaluating Cetuximab in Combination With Mitoxantrone Plus Prednisone After Docetaxel for Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2012, 10, 6–14. [Google Scholar] [CrossRef]
- Agus, D.B.; Sweeney, C.J.; Morris, M.J.; Mendelson, D.S.; McNeel, D.G.; Ahmann, F.R.; Wang, J.; Derynck, M.K.; Ng, K.; Lyons, B.; et al. Efficacy and Safety of Single-Agent Pertuzumab (RhuMAb 2C4), a Human Epidermal Growth Factor Receptor Dimerization Inhibitor, in Castration-Resistant Prostate Cancer After Progression from Taxane-Based Therapy. J. Clin. Oncol. 2007, 25, 675–681. [Google Scholar] [CrossRef]
- Ziada, A.; Barqawi, A.; Glode, L.M.; Varella-Garcia, M.; Crighton, F.; Majeski, S.; Rosenblum, M.; Kane, M.; Chen, L.; Crawford, E.D. The Use of Trastuzumab in the Treatment of Hormone Refractory Prostate Cancer; Phase II Trial. Prostate 2004, 60, 332–337. [Google Scholar] [CrossRef]
- Morris, M.J.; Reuter, V.E.; Kelly, W.K.; Slovin, S.F.; Kenneson, K.; Verbel, D.; Osman, I.; Scher, H.I. HER-2 Profiling and Targeting in Prostate Carcinoma: A Phase II Trial of Trastuzumab Alone and with Paclitaxel. Cancer 2002, 94, 980–986. [Google Scholar] [CrossRef]
- Lara, P.N.; Chee, K.G.; Longmate, J.; Ruel, C.; Meyers, F.J.; Gray, C.R.; Edwards, R.G.; Gumerlock, P.H.; Twardowski, P.; Doroshow, J.H.; et al. Trastuzumab plus Docetaxel in HER-2/Neu-Positive Prostate Carcinoma: Final Results from the California Cancer Consortium Screening and Phase II Trial. Cancer 2004, 100, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Ching, J.B.; Dahut, W.L. VEGF Inhibitors and Prostate Cancer Therapy. Curr. Mol. Pharmacol. 2009, 2, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Trial Comparing Docetaxel and Prednisone With or Without Bevacizumab in Men With Metastatic Castration-Resistant Prostate Cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Reyno, L.; Venner, P.M.; Ernst, S.D.; Moore, M.; Geary, R.S.; Chi, K.; Hall, S.; Walsh, W.; Dorr, A.; et al. A Randomized Phase II and Pharmacokinetic Study of the Antisense Oligonucleotides ISIS 3521 and ISIS 5132 in Patients with Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2002, 8, 2530–2535. [Google Scholar] [PubMed]
- Dahut, W.L.; Scripture, C.; Posadas, E.; Jain, L.; Gulley, J.L.; Arlen, P.M.; Wright, J.J.; Yu, Y.; Cao, L.; Steinberg, S.M.; et al. A Phase II Clinical Trial of Sorafenib in Androgen-Independent Prostate Cancer. Clin. Cancer Res. 2008, 14, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Aragon-Ching, J.B.; Jain, L.; Gulley, J.L.; Arlen, P.M.; Wright, J.J.; Steinberg, S.M.; Draper, D.; Venitz, J.; Jones, E.; Chen, C.C.; et al. Final Analysis of a Phase II Trial Using Sorafenib for Metastatic Castration-Resistant Prostate Cancer. BJU Int. 2009, 103, 1636–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezaro, C.; Rosenthal, M.A.; Gurney, H.; Davis, I.D.; Underhill, C.; Boyer, M.J.; Kotasek, D.; Solomon, B.; Toner, G.C. An Open-Label, Single-Arm Phase Two Trial of Gefitinib in Patients With Advanced or Metastatic Castration-Resistant Prostate Cancer. Am. J. Clin. Oncol. 2009, 32, 338–341. [Google Scholar] [CrossRef]
- Dahut, W.L.; Madan, R.A.; Karakunnel, J.J.; Adelberg, D.; Gulley, J.L.; Turkbey, I.B.; Chau, C.H.; Spencer, S.D.; Mulquin, M.; Wright, J.; et al. Phase II Clinical Trial of Cediranib in Patients with Metastatic Castration-Resistant Prostate Cancer: Phase II Cediranib in Advanced Prostate Cancer. BJU Int. 2013, 111, 1269–1280. [Google Scholar] [CrossRef]
- Heath, E.; Heilbrun, L.; Mannuel, H.; Liu, G.; Lara, P.; Monk, J.P.; Flaig, T.; Zurita, A.; Mack, P.; Vaishampayan, U.; et al. Phase II, Multicenter, Randomized Trial of Docetaxel plus Prednisone with or Without Cediranib in Men with Chemotherapy-Naive Metastatic Castrate-Resistant Prostate Cancer. Oncologist 2019, 24, 1149. [Google Scholar] [CrossRef] [Green Version]
- Barata, P.C.; Cooney, M.; Mendiratta, P.; Gupta, R.; Dreicer, R.; Garcia, J.A. Phase I/II Study Evaluating the Safety and Clinical Efficacy of Temsirolimus and Bevacizumab in Patients with Chemotherapy Refractory Metastatic Castration-Resistant Prostate Cancer. Invest. New Drugs 2019, 37, 331–337. [Google Scholar] [CrossRef]
- Picus, J.; Halabi, S.; Kelly, W.K.; Vogelzang, N.J.; Whang, Y.E.; Kaplan, E.B.; Stadler, W.M.; Small, E.J.; Cancer and Leukemia Group B. A Phase 2 Study of Estramustine, Docetaxel, and Bevacizumab in Men with Castrate-Resistant Prostate Cancer: Results from Cancer and Leukemia Group B Study 90006. Cancer 2011, 117, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Kim, H.S.; Park, S.H.; Kim, B.-S.; Kim, K.H.; Lee, H.J.; Song, H.S.; Shin, D.-Y.; Lee, H.Y.; Kim, H.-G.; et al. Phase II Study of Dovitinib in Patients with Castration-Resistant Prostate Cancer (KCSG-GU11-05). Cancer Res. Treat. 2018, 50, 1252–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, S.S.; Joshua, A.M.; Gregg, R.; Booth, C.M.; Murray, N.; Golubovic, J.; Wang, L.; Harris, P.; Chi, K.N. A Phase II Study of GW786034 (Pazopanib) With or Without Bicalutamide in Patients With Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2015, 13, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus Placebo in Combination with Docetaxel and Prednisone for Treatment of Men with Metastatic Castration-Resistant Prostate Cancer (VENICE): A Phase 3, Double-Blind Randomised Trial. Lancet Oncol. 2013, 14, 760–768. [Google Scholar] [CrossRef]
- Stadler, W.M.; Cao, D.; Vogelzang, N.J.; Ryan, C.W.; Hoving, K.; Wright, R.; Karrison, T.; Vokes, E.E. A Randomized Phase II Trial of the Antiangiogenic Agent SU5416 in Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2004, 10, 3365–3370. [Google Scholar] [CrossRef] [Green Version]
- Figg, W.D.; Dahut, W.; Duray, P.; Hamilton, M.; Tompkins, A.; Steinberg, S.M.; Jones, E.; Premkumar, A.; Linehan, W.M.; Floeter, M.K.; et al. A Randomized Phase II Trial of Thalidomide, an Angiogenesis Inhibitor, in Patients with Androgen-Independent Prostate Cancer. Clin. Cancer Res. 2001, 7, 1888–1893. [Google Scholar]
- Drake, M.J.; Robson, W.; Mehta, P.; Schofield, I.; Neal, D.E.; Leung, H.Y. An Open-Label Phase II Study of Low-Dose Thalidomide in Androgen-Independent Prostate Cancer. Br. J. Cancer 2003, 88, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing Itraconazole as a Treatment for Advanced Prostate Cancer: A Noncomparative Randomized Phase II Trial in Men with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef]
- Albanell, J.; Rojo, F.; Averbuch, S.; Feyereislova, A.; Mascaro, J.M.; Herbst, R.; LoRusso, P.; Rischin, D.; Sauleda, S.; Gee, J.; et al. Pharmacodynamic Studies of the Epidermal Growth Factor Receptor Inhibitor ZD1839 in Skin from Cancer Patients: Histopathologic and Molecular Consequences of Receptor Inhibition. J. Clin. Oncol. 2002, 20, 110–124. [Google Scholar] [CrossRef]
- Pin, E.; Stratton, S.; Belluco, C.; Liotta, L.; Nagle, R.; Hodge, K.A.; Deng, J.; Dong, T.; Baldelli, E.; Petricoin, E.; et al. A Pilot Study Exploring the Molecular Architecture of the Tumor Microenvironment in Human Prostate Cancer Using Laser Capture Microdissection and Reverse Phase Protein Microarray. Mol. Oncol. 2016, 10, 1585–1594. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Kulik, G. Signaling Pathways That Control Apoptosis in Prostate Cancer. Cancers 2021, 13, 937. https://doi.org/10.3390/cancers13050937
Ali A, Kulik G. Signaling Pathways That Control Apoptosis in Prostate Cancer. Cancers. 2021; 13(5):937. https://doi.org/10.3390/cancers13050937
Chicago/Turabian StyleAli, Amaal, and George Kulik. 2021. "Signaling Pathways That Control Apoptosis in Prostate Cancer" Cancers 13, no. 5: 937. https://doi.org/10.3390/cancers13050937
APA StyleAli, A., & Kulik, G. (2021). Signaling Pathways That Control Apoptosis in Prostate Cancer. Cancers, 13(5), 937. https://doi.org/10.3390/cancers13050937