
Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma



Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
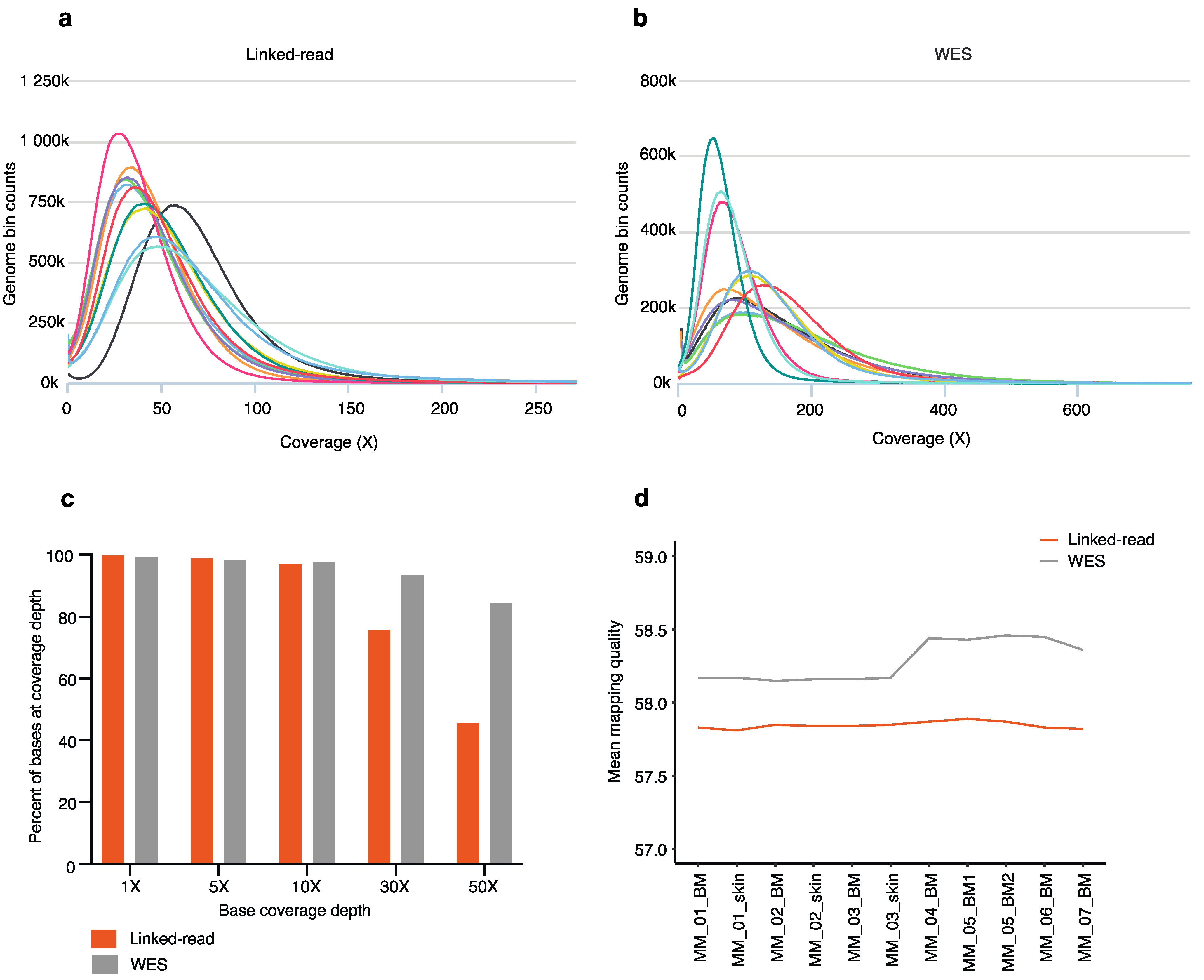
2.1. Sequencing and Mapping Quality Statistics
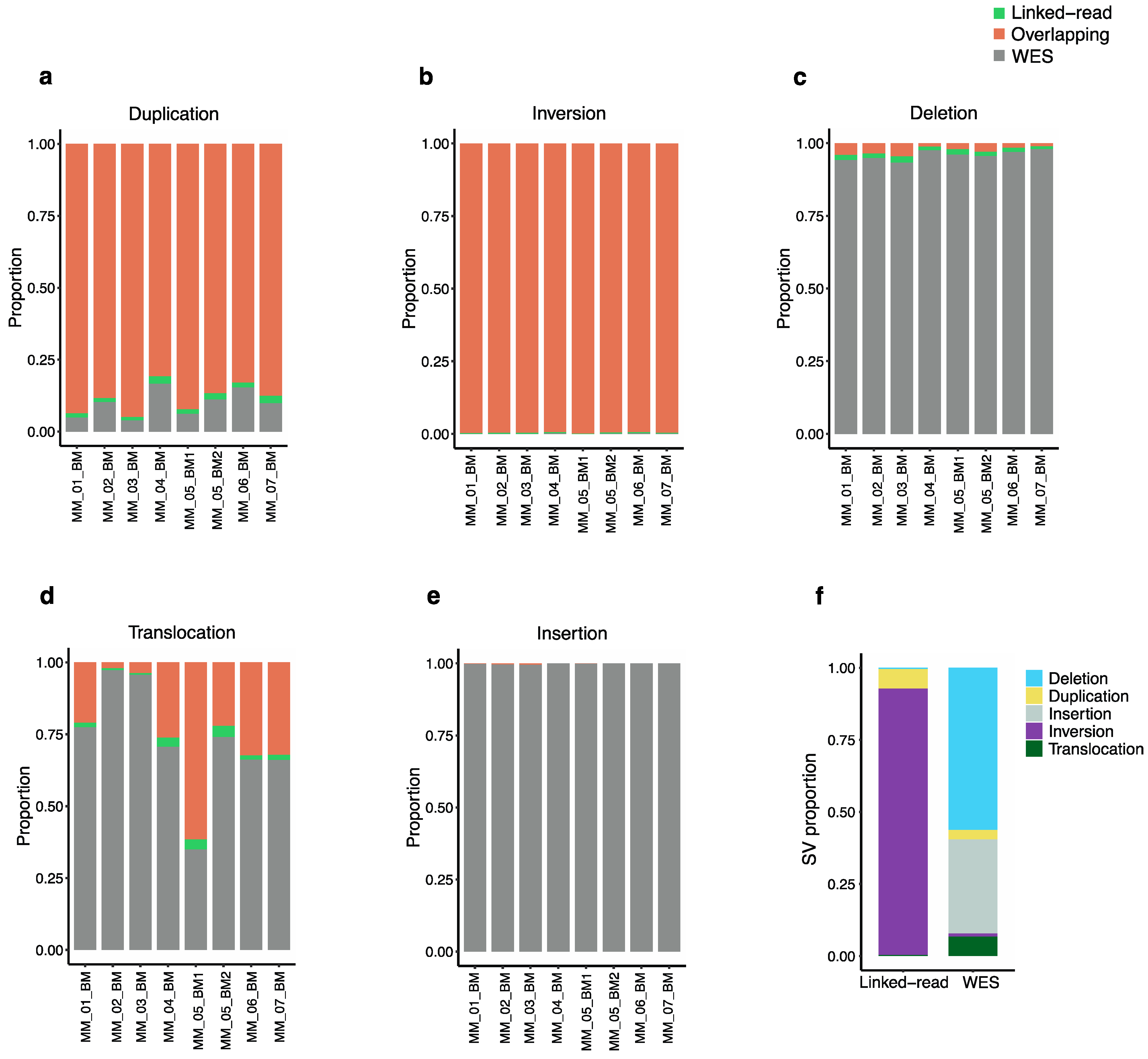
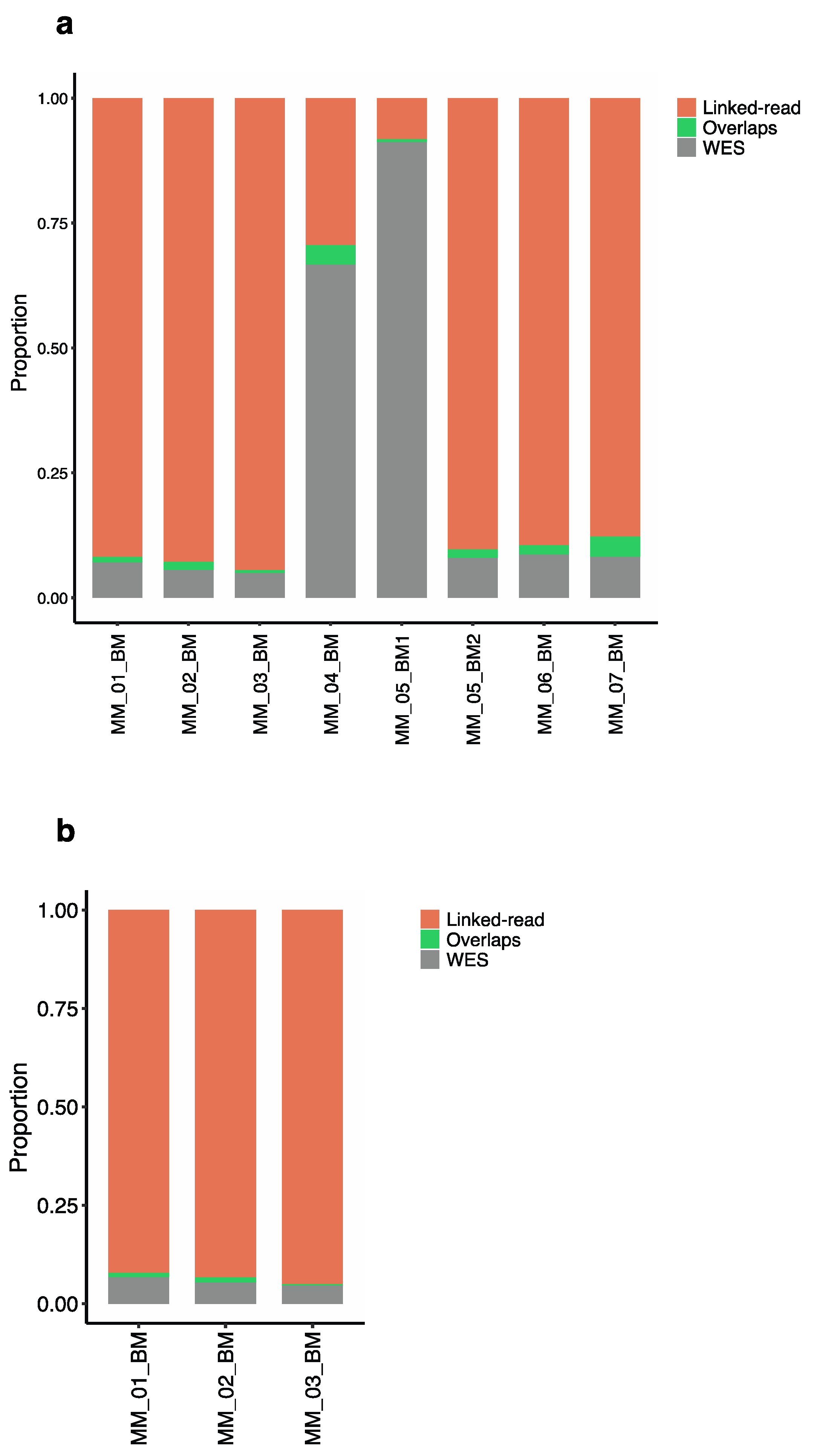
2.2. Detection of Total SVs by Linked-Read Sequencing and WES
2.3. Detection of Somatic SVs by Linked-Read Sequencing and WES
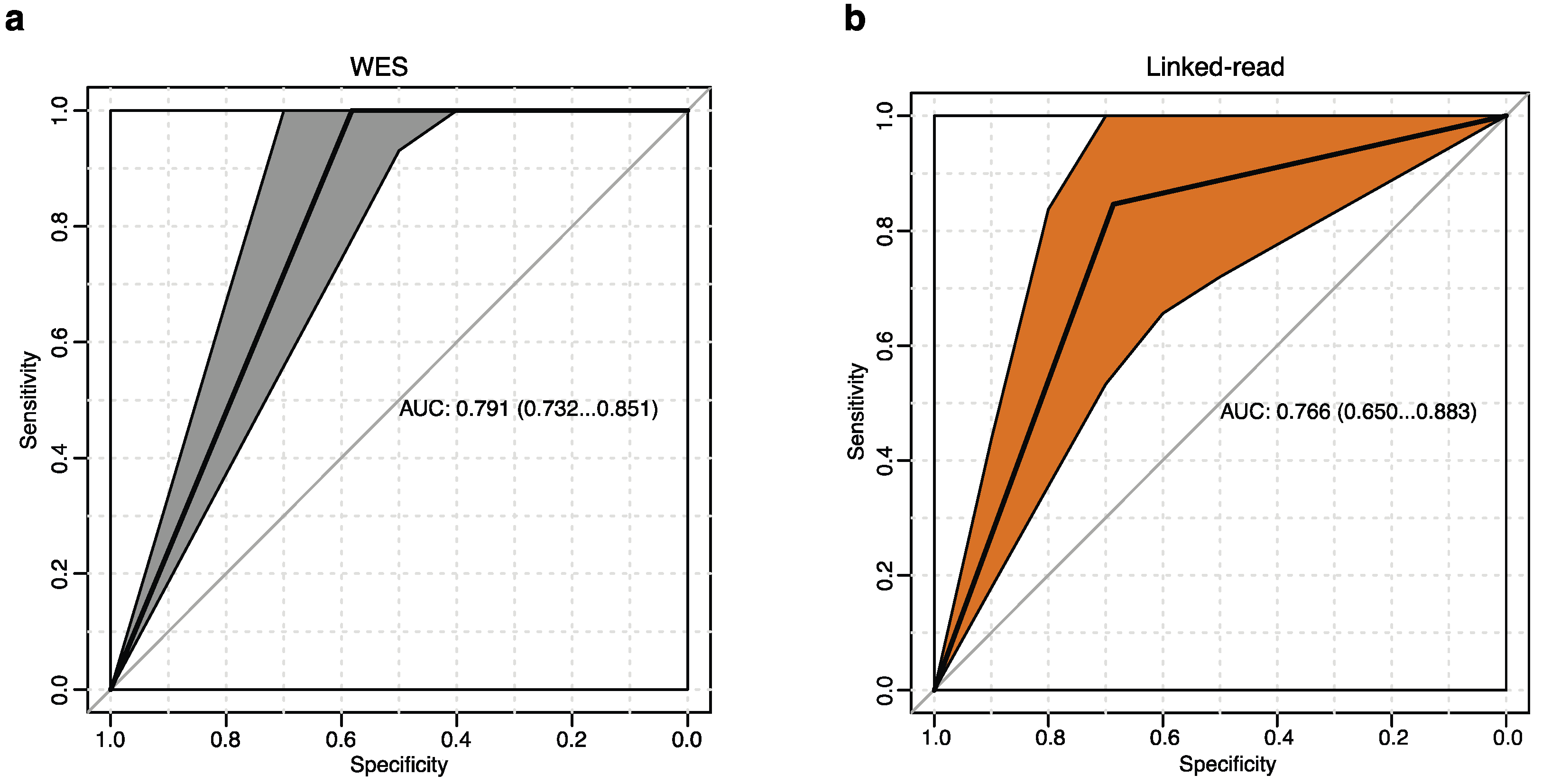
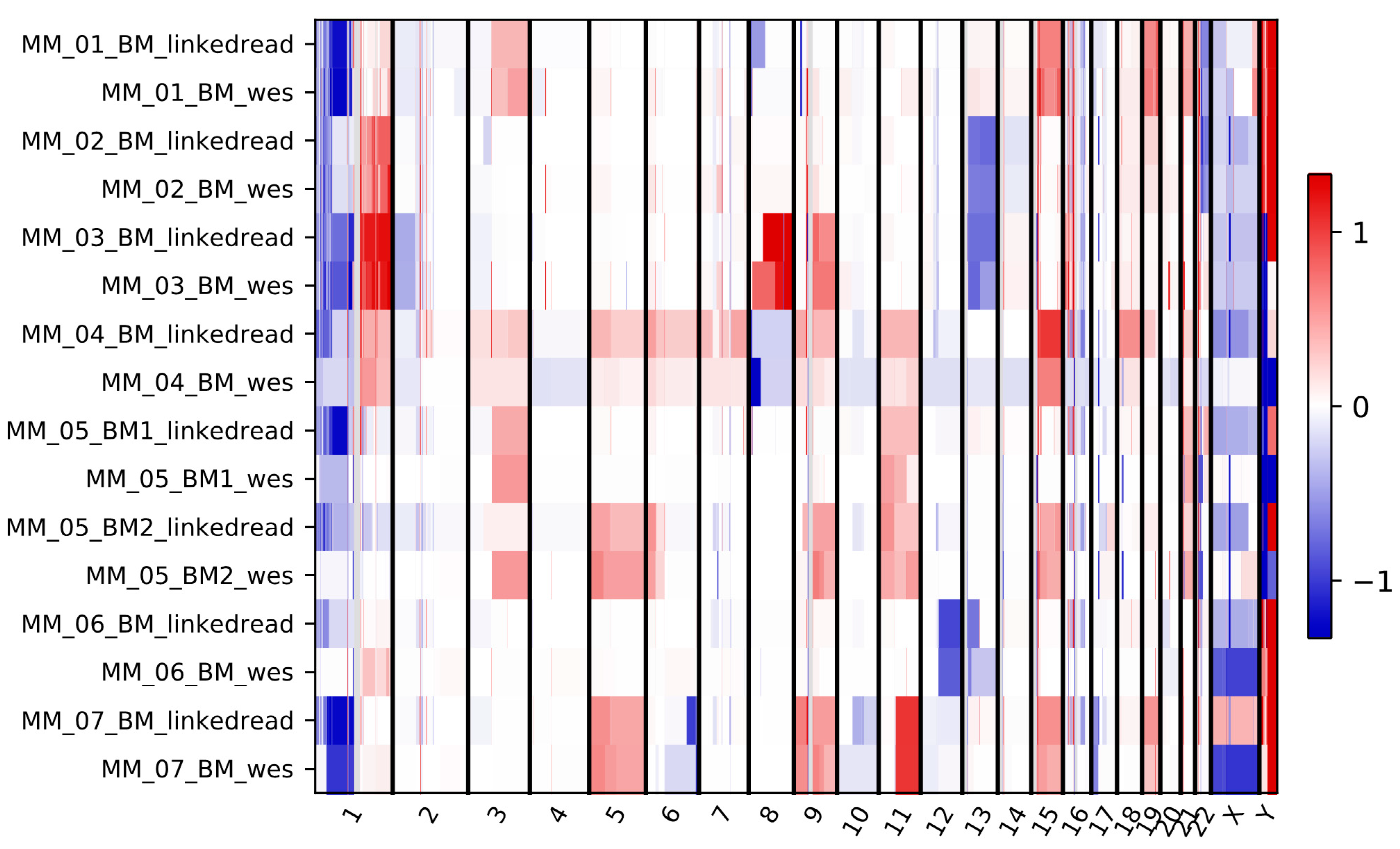
2.4. Performance Evaluation for Detecting Known Clinical Cytogenetic Alterations
2.5. Performance Evaluation for Detecting MM-Specific SV Hotspots
2.6. Detection of Total Short Variants by Linked-Read Sequencing and WES
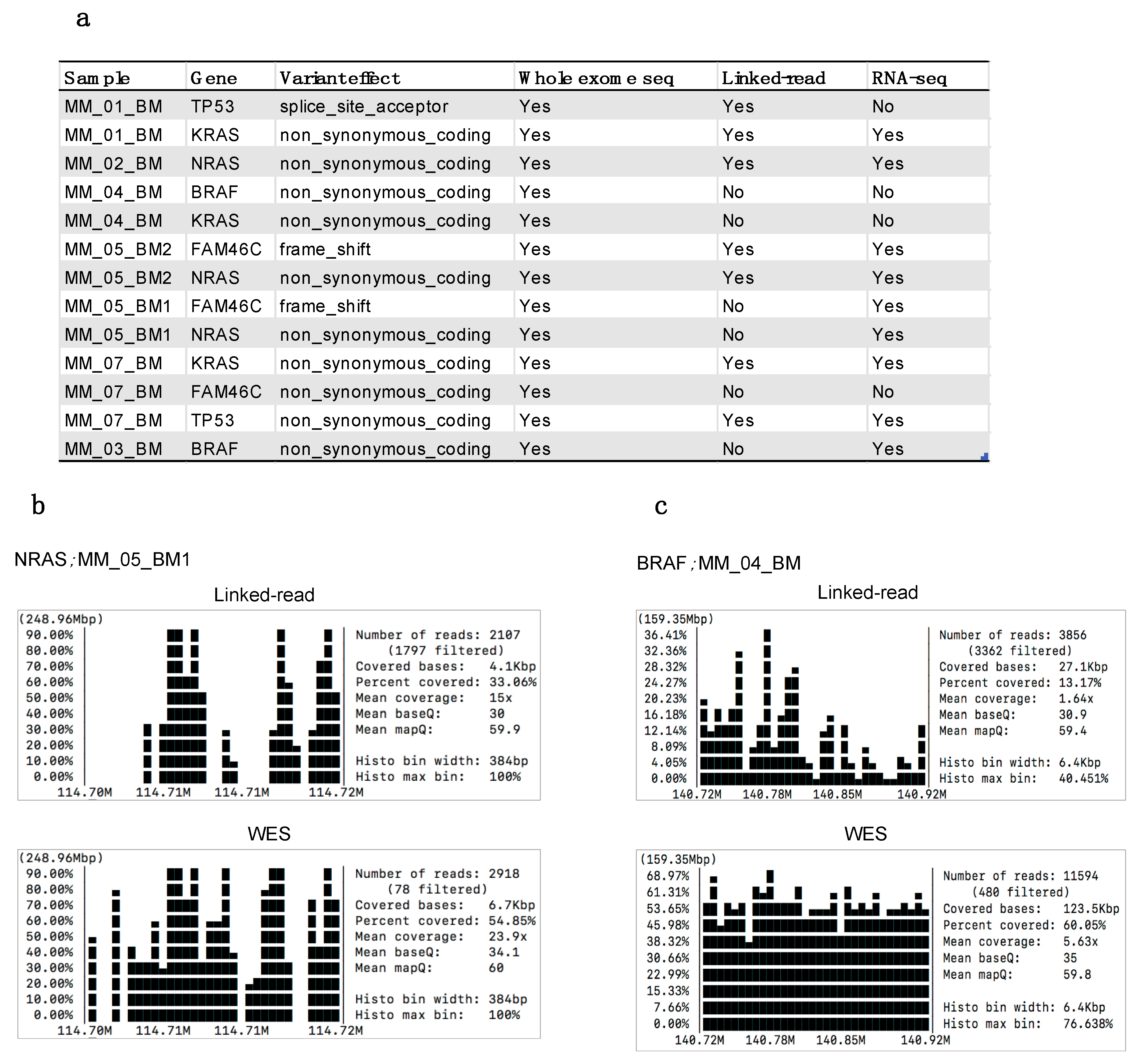
2.7. Performance Evaluation for Detecting Myeloma Specific Mutations
2.8. Comparison of Short Variants and SVs Detected by Linked-Read Sequencing, WES and RNA-seq
3. Discussion
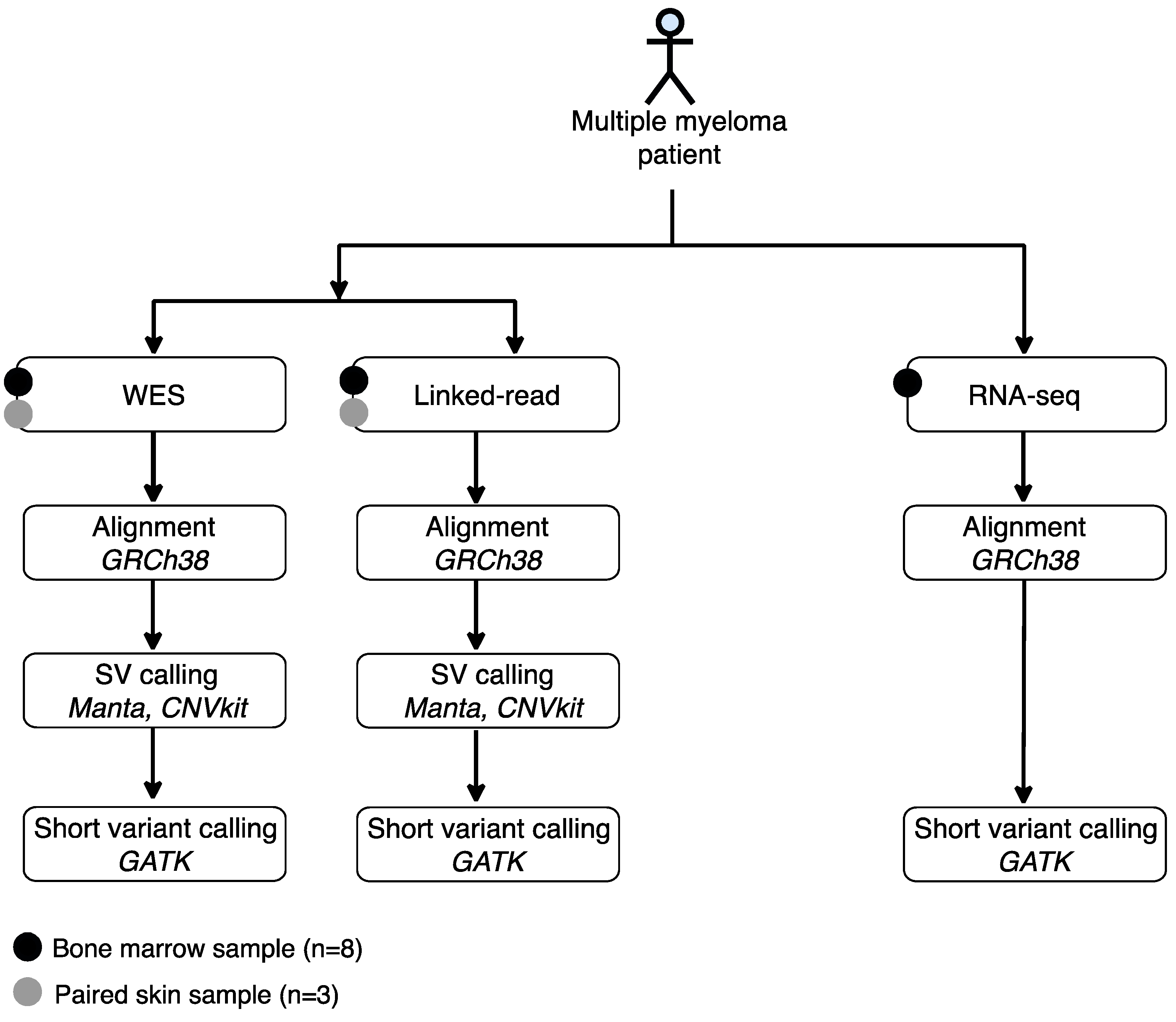
4. Materials and Methods
4.1. Patient Materials and Ethical Compliance
4.2. Sample Processing
4.3. Whole-Exome Sequencing (WES)
4.4. Linked-Read Exome Sequencing
4.5. RNA Sequencing
4.6. Sequencing and Mapping Quality Statistics
4.7. Short Variant Calling
4.8. Structural Variant (SV) Calling
4.9. Overlapping Variants
4.10. Somatic Mutation Overlap Analysis
4.11. Clinically Relevant Alterations
4.12. Sensitivity and Specificity Calculation
4.13. Identification of SV Hotspots
4.14. Identification of Copy Number Variants (CNVs)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, M. Structural variation: The genome’s hidden architecture. Nat. Methods 2012, 9, 133–137. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. Author Correction: The landscape of genomic alterations across childhood cancers. Nature 2018, 559, E10. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; Imielinski, M. Modeling cancer rearrangement landscapes. Curr. Opin. Syst. Biol. 2017, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Rustad, E.H.; Yellapantula, V.D.; Glodzik, D.; Maclachlan, K.H.; Diamond, B.; Boyle, E.M.; Ashby, C.; Blaney, P.; Gundem, G.; Hultcrantz, M.; et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov. 2020, 1, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huddleston, J.; Chaisson, M.J.P.; Steinberg, K.M.; Warren, W.; Hoekzema, K.; Gordon, D.; Graves-Lindsay, T.A.; Munson, K.M.; Kronenberg, Z.N.; Vives, L.; et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2018, 28, 144. [Google Scholar] [CrossRef] [Green Version]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [Green Version]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Wala, J.A.; Bandopadhayay, P.; Greenwald, N.F.; O’Rourke, R.; Sharpe, T.; Stewart, C.; Schumacher, S.; Li, Y.; Weischenfeldt, J.; Yao, X.; et al. SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018, 28, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Horak, P.; Fröhling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Shashikant, C.S.; Jensen, M.; Altman, N.S.; Girirajan, S. Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non-uniformity. Sci. Rep. 2017, 7, 885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef] [Green Version]
- Huddleston, J.; Eichler, E.E. An Incomplete Understanding of Human Genetic Variation. Genetics 2016, 202, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Steyaert, W.; Callens, S.; Coucke, P.; Dermaut, B.; Hemelsoet, D.; Terryn, W.; Poppe, B. Future perspectives of genome-scale sequencing. Acta Clin. Belg. 2018, 73, 7–10. [Google Scholar] [CrossRef]
- Amarasinghe, K.C.; Li, J.; Hunter, S.M.; Ryland, G.L.; Cowin, P.A.; Campbell, I.G.; Halgamuge, S.K. Inferring copy number and genotype in tumour exome data. BMC Genom. 2014, 15, 732. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [Green Version]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Petrov, D.A.; Fiston-Lavier, A.S. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 2014, 9, e106689. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Schnable, J.C.; Yeh, C.T.; Wu, L.; Liu, C.; Hu, H.C.; Dalgard, C.L.; Sarkar, S.; Schnable, P.S. Linked read technology for assembling large complex and polyploid genomes. BMC Genom. 2018, 19, 651. [Google Scholar] [CrossRef] [PubMed]
- Uguen, K.; Jubin, C.; Duffourd, Y.; Bardel, C.; Malan, V.; Dupont, J.M.; El Khattabi, L.; Chatron, N.; Vitobello, A.; Rollat-Farnier, P.A.; et al. Genome sequencing in cytogenetics: Comparison of short-read and linked-read approaches for germline structural variant detection and characterization. Mol. Genet. Genom. Med. 2020, 8, e1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyanow, R.; Wu, H.T.; Raphael, B.J. Identifying structural variants using linked-read sequencing data. Bioinformatics 2018, 34, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, S.U.; Nadauld, L.D.; Lau, B.T.; Chen, J.; Wood-Bouwens, C.; Ford, J.M.; Kuo, C.J.; Ji, H.P. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Med. 2017, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A.; et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Mansfield, E.; Pazdur, R. Next-Generation Sequencing in Oncology in the Era of Precision Medicine. JAMA Oncol. 2016, 2, 13–14. [Google Scholar] [CrossRef]
- Hood, L.; Rowen, L. The Human Genome Project: Big science transforms biology and medicine. Genome Med. 2013, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Nordlund, J.; Marincevic-Zuniga, Y.; Cavelier, L.; Raine, A.; Martin, T.; Lundmark, A.; Abrahamsson, J.; Norén-Nyström, U.; Lönnerholm, G.; Syvänen, A.C. Refined detection and phasing of structural aberrations in pediatric acute lymphoblastic leukemia by linked-read whole-genome sequencing. Sci. Rep. 2020, 10, 2512. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Kao, C.; Gonzalez, M.V.; Mafra, F.A.; Pellegrino da Silva, R.; Li, M.; Wenzel, S.S.; Wimmer, K.; Hakonarson, H.; Wang, K. LinkedSV for detection of mosaic structural variants from linked-read exome and genome sequencing data. Nat. Commun. 2019, 10, 5585. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.M.; Silvennoinen, R.; Anttila, P.; Tamborero, D.; Eldfors, S.; Yadav, B.; Karjalainen, R.; Kuusanmäki, H.; Lievonen, J.; Parsons, A.; et al. Identification of precision treatment strategies for relapsed/refractory multiple myeloma by functional drug sensitivity testing. Oncotarget 2017, 8, 56338–56350. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmäki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef]
- Kumar, A.; Kankainen, M.; Parsons, A.; Kallioniemi, O.; Mattila, P.; Heckman, C.A. The impact of RNA sequence library construction protocols on transcriptomic profiling of leukemia. BMC Genom. 2017, 18, 629. [Google Scholar] [CrossRef] [Green Version]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Auer, P.L.; Reiner, A.P.; Wang, G.; Kang, H.M.; Abecasis, G.R.; Altshuler, D.; Bamshad, M.J.; Nickerson, D.A.; Tracy, R.P.; Rich, S.S.; et al. Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am. J. Hum. Genet. 2016, 99, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Eberle, M.A.; Fritzilas, E.; Krusche, P.; Källberg, M.; Moore, B.L.; Bekritsky, M.A.; Iqbal, Z.; Chuang, H.Y.; Humphray, S.J.; Halpern, A.L.; et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017, 27, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Gender | Age at Diagnosis | Disease Status | Myeloma Characteristics | ISS Stage | WES | Linked-Read | RNA-Seq | Sample Type | Cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|
| MM_01_03 | Male | 57 | Relapse | IgG lambda | 3 | Yes | Yes | Yes | Bone marrow and skin | del(13q), 1p loss |
| MM_02_03 | Male | 65 | Relapse | IgG kappa | 2 | Yes | Yes | Yes | Bone marrow and skin | del (13q), possibly del(14) |
| MM_03_03 | Male | 60 | Relapse | IgA lambda | 3 | Yes | Yes | Yes | Bone marrow and skin | del (13q), t(4;14), gain(1q), 14q32 |
| MM_04_06 | Female | 69 | Relapse | IgA, kappa | 1 | Yes | Yes | Yes | Bone marrow | gain (1q), trisomy 9, trisomy 11, Other deviation: trisomy 5, trisomy 15 |
| MM_05_03 | Male | 56 | Relapse | Unknown, kappa | 1 | Yes | Yes | Yes | Bone marrow | trisomy 9, trisomy 11, other deviation: trisomy 5, trisomy 15 |
| MM_05_09 | Male | 56 | Relapse | Unknown, kappa | 1 | Yes | Yes | Yes | Bone marrow | trisomy 9, trisomy 11, other deviation: trisomy 5, trisomy 15 |
| MM_06_03 | Male | 41 | Refractory | IgG lambda | ND | Yes | Yes | Yes | Bone marrow | Monosomy 13, del(13q) |
| MM_07_03 | Male | 56 | Relapse | IgG kappa | ND | Yes | Yes | Yes | Bone marrow | del (17p), gain (1q), 1p36 loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Adhikari, S.; Kankainen, M.; Heckman, C.A. Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma. Cancers 2021, 13, 1212. https://doi.org/10.3390/cancers13061212
Kumar A, Adhikari S, Kankainen M, Heckman CA. Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma. Cancers. 2021; 13(6):1212. https://doi.org/10.3390/cancers13061212
Chicago/Turabian StyleKumar, Ashwini, Sadiksha Adhikari, Matti Kankainen, and Caroline A. Heckman. 2021. "Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma" Cancers 13, no. 6: 1212. https://doi.org/10.3390/cancers13061212
APA StyleKumar, A., Adhikari, S., Kankainen, M., & Heckman, C. A. (2021). Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma. Cancers, 13(6), 1212. https://doi.org/10.3390/cancers13061212