
Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
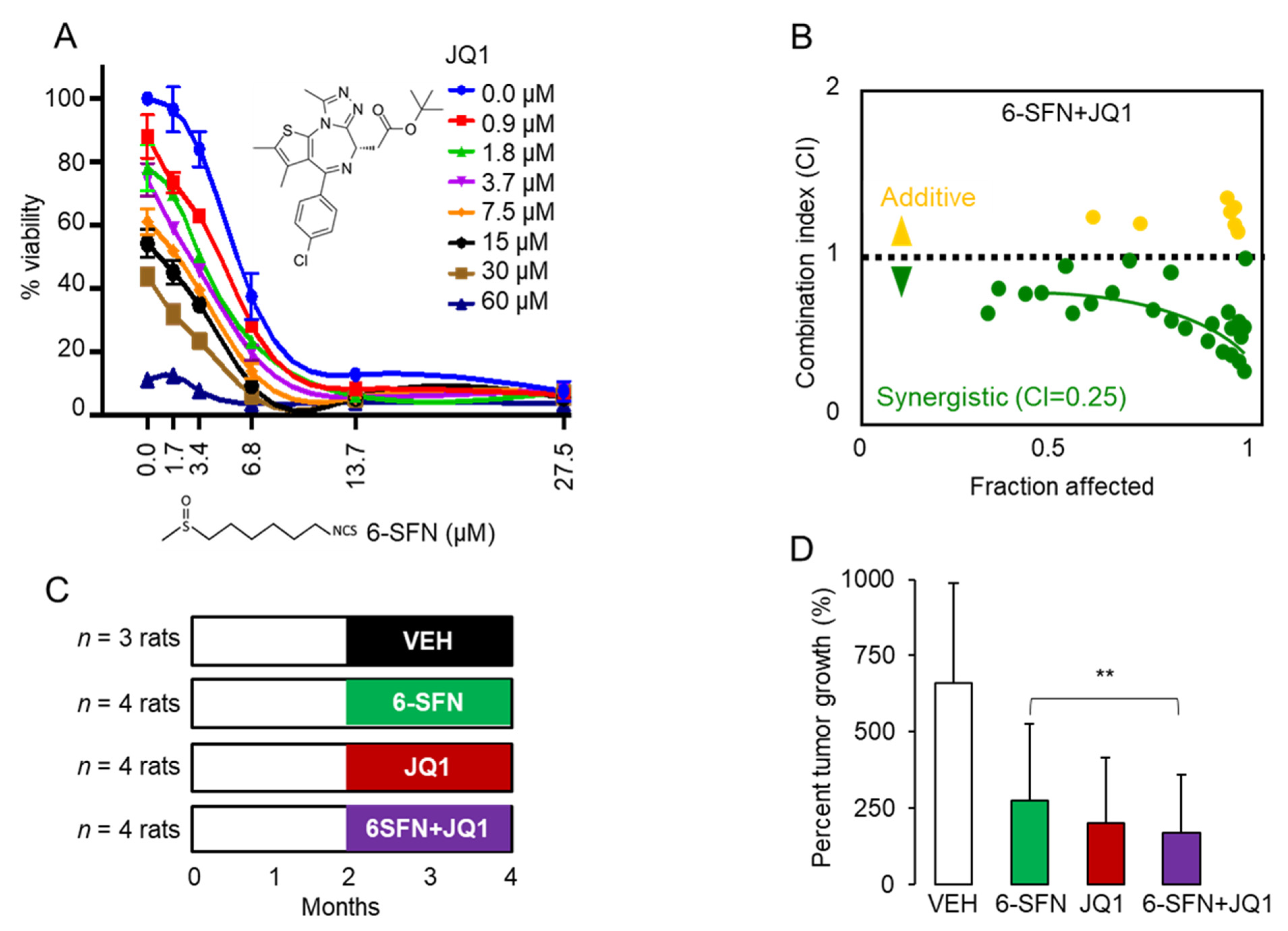
2.1. 6-SFN + JQ1 Act Synergistically in Human Colon Cancer Cells and Suppress Colon Polyps In Vivo
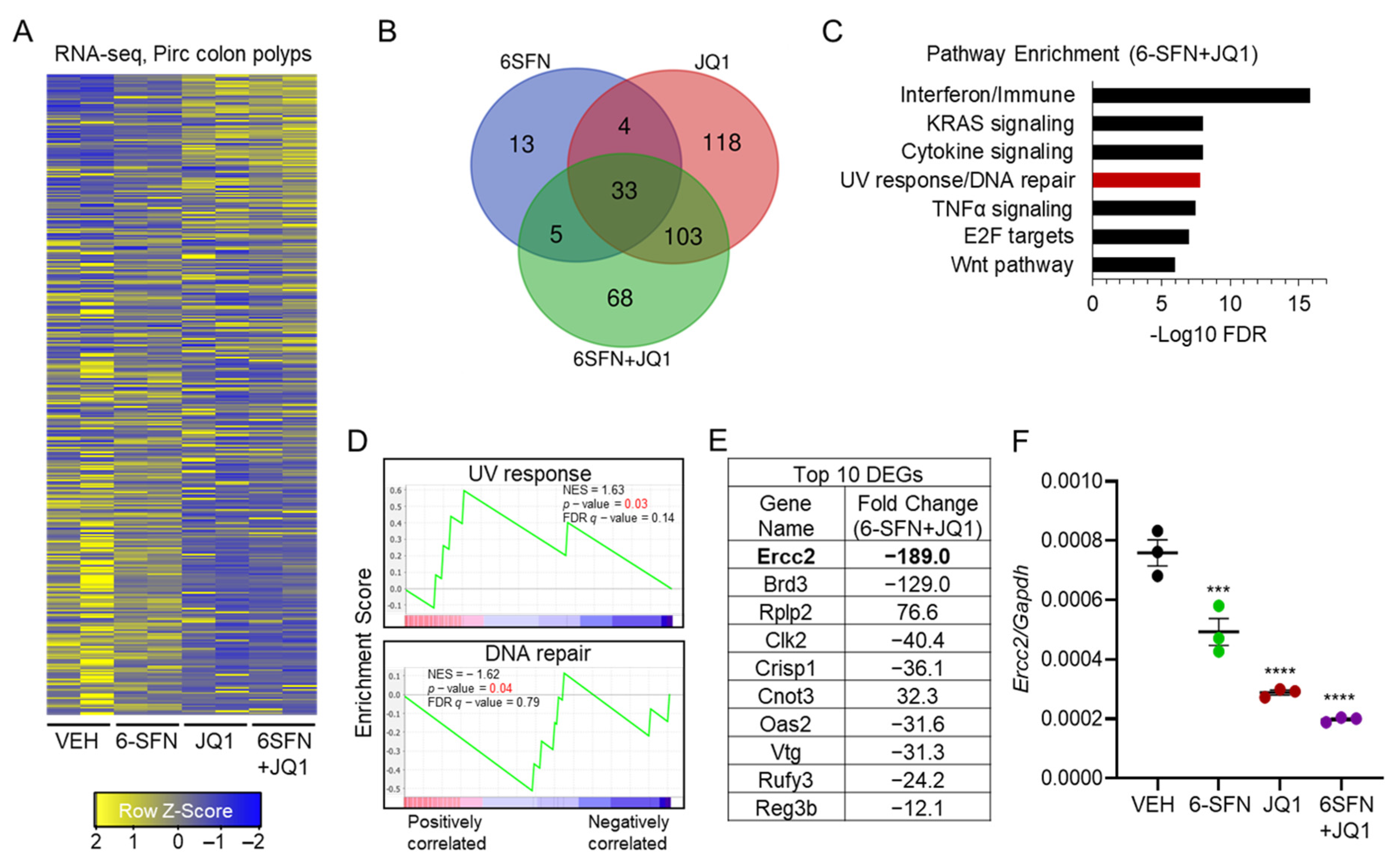
2.2. Transcriptomics Prioritizes Ercc2 as a Key ‘Synergy/Cooperativity’ Gene in Pirc Colon Tumors
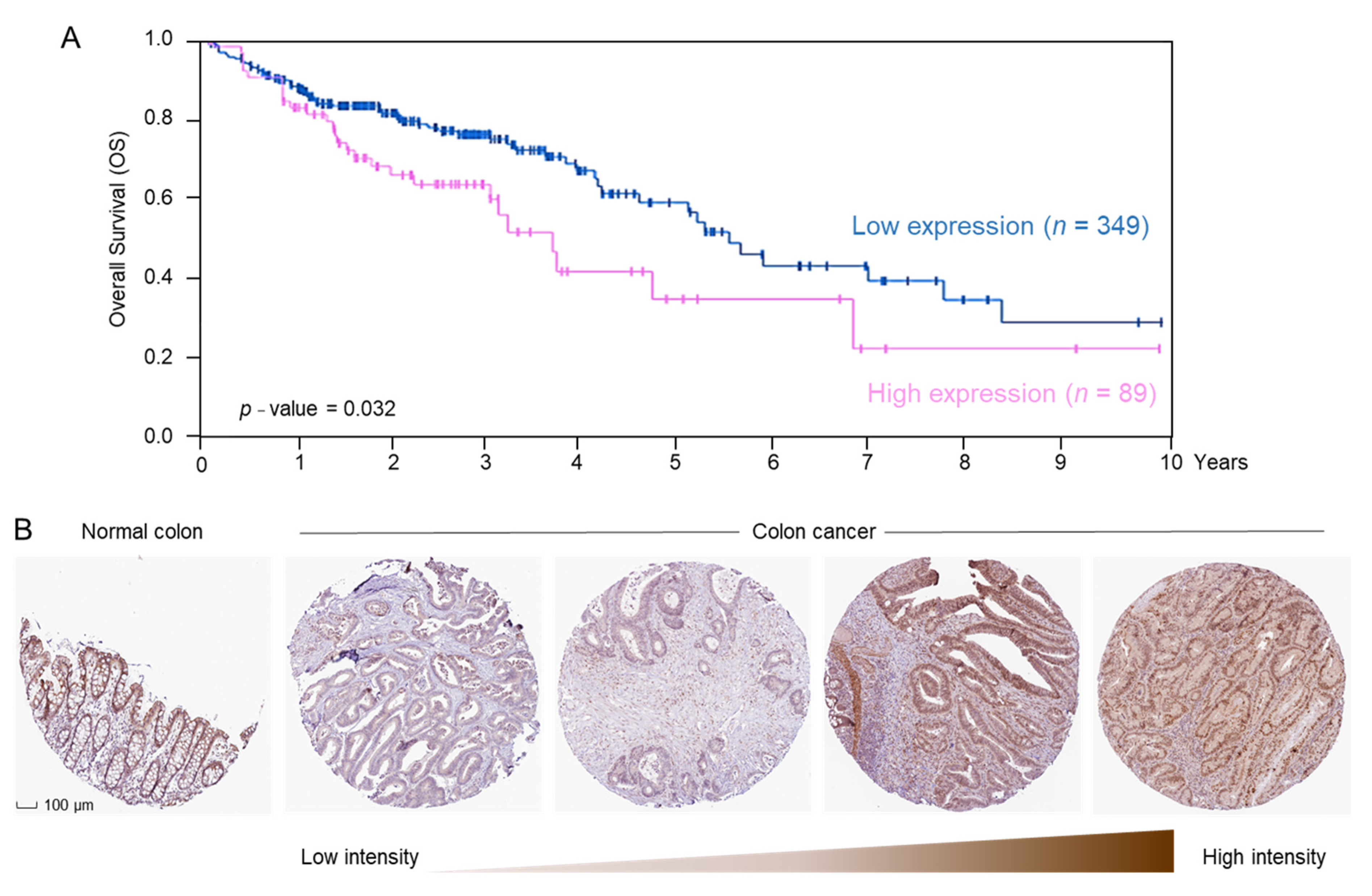
2.3. ERCC2 Overexpression Is Associated with Reduced Survival in CRC Patients
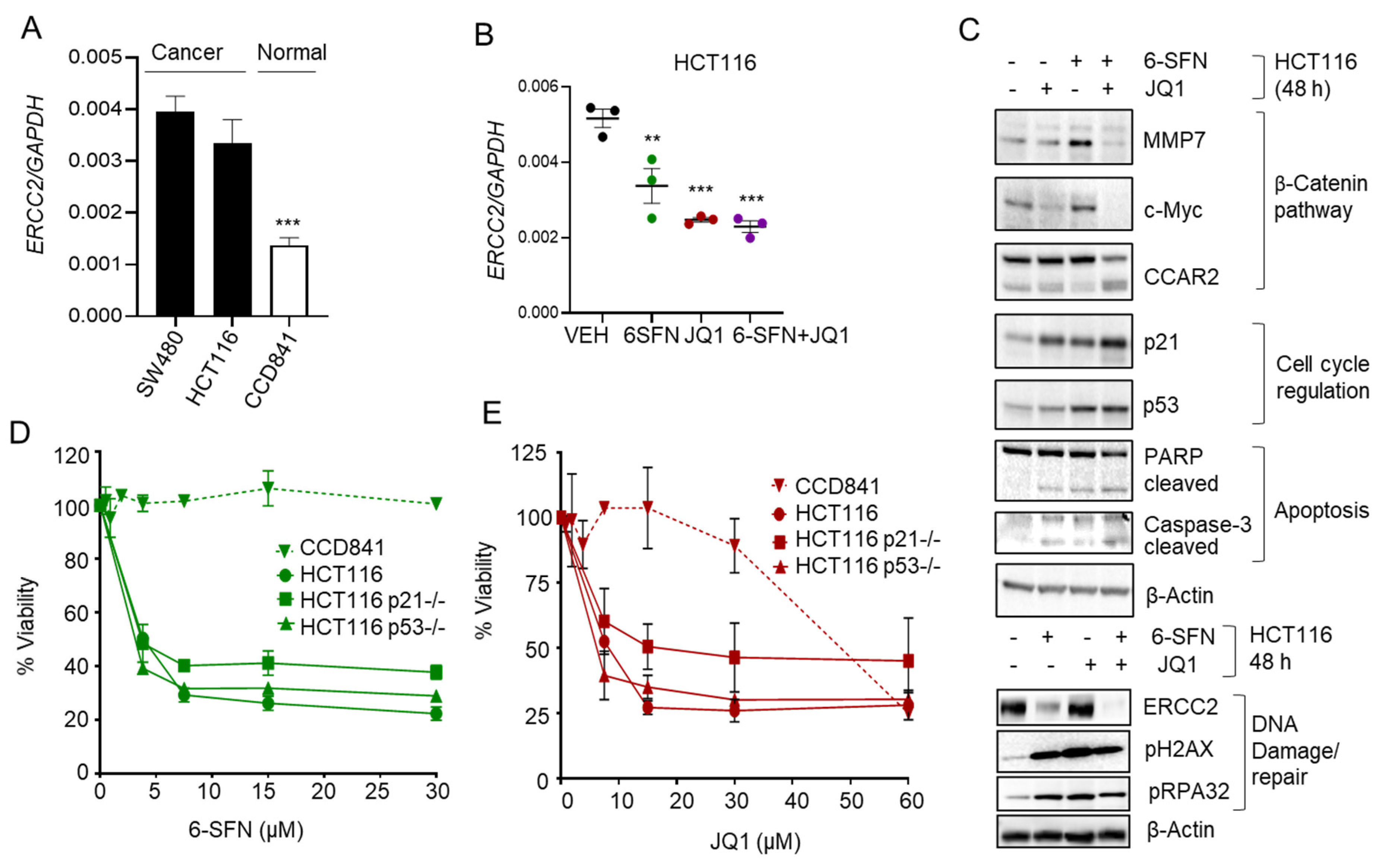
2.4. 6-SFN + JQ1 Co-Treatment Downregulates ERCC2 in Human Colon Cancer Cells
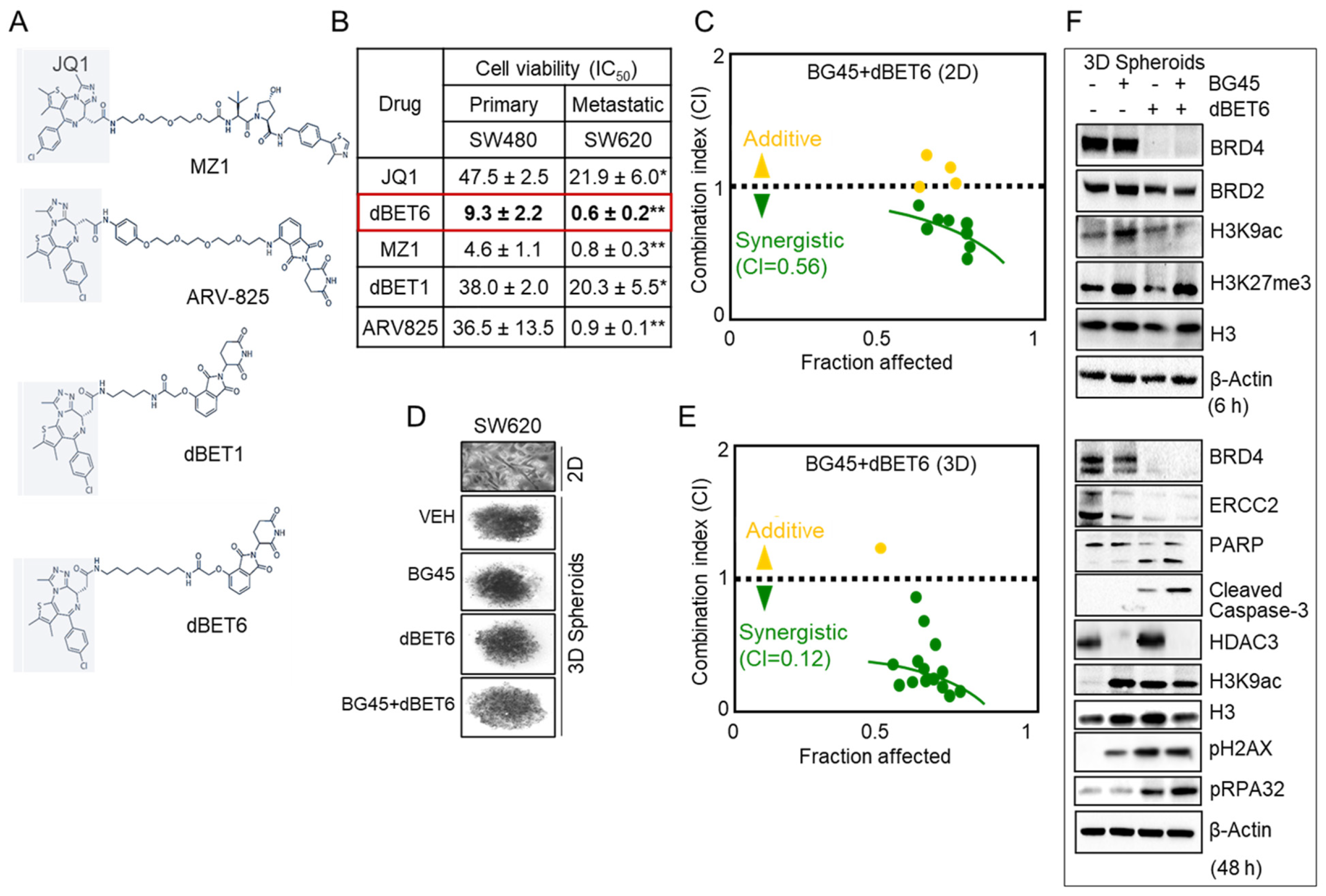
2.5. HDAC3 Inhibitor Plus BET Degrader Co-Treatment as Second-Generation Epigenetic Therapy
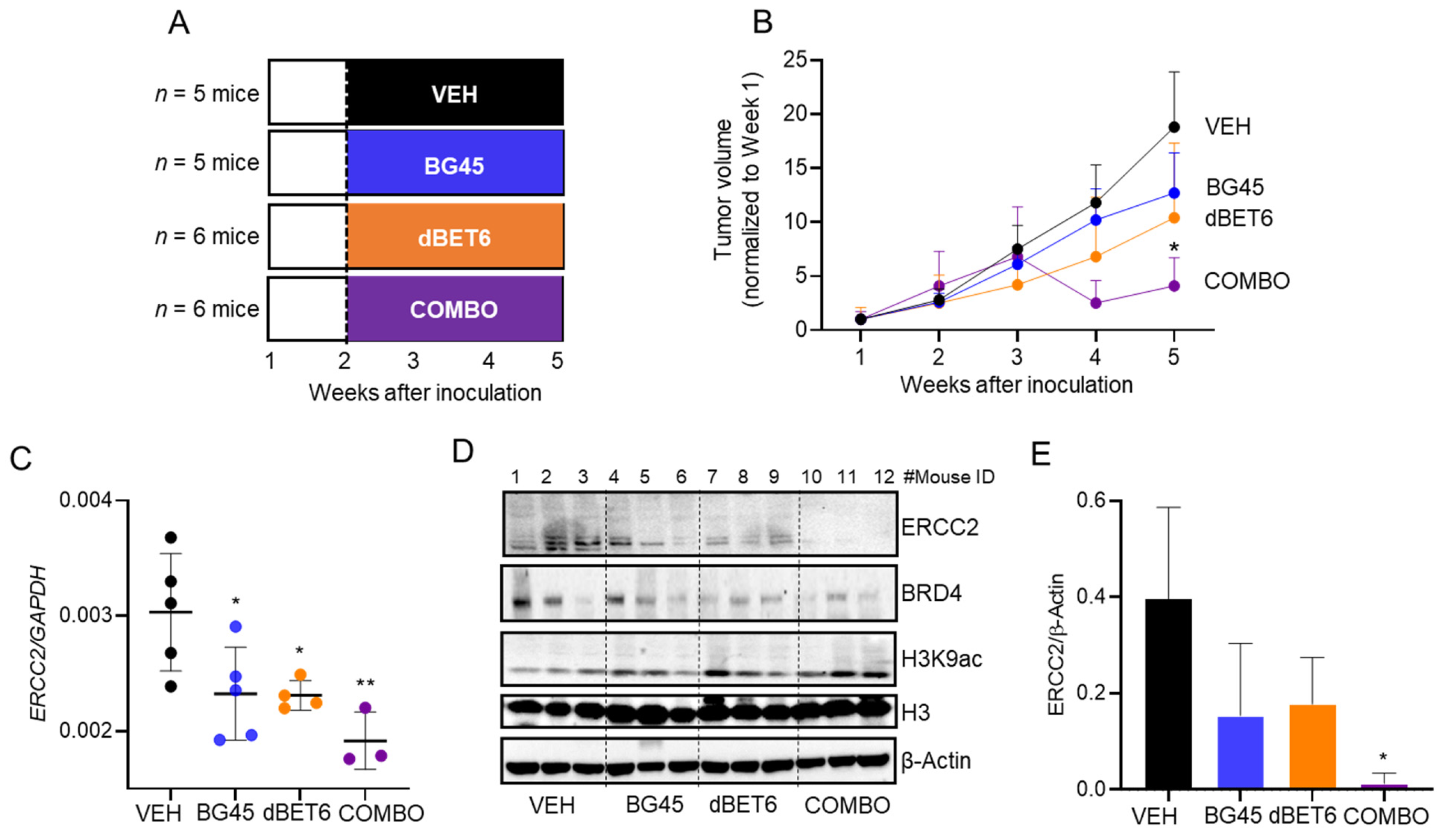
2.6. Antitumor Activity of dBET6 + BG45 in SW620 Xenografts
3. Discussion
4. Materials and Methods
4.1. Cells and Treatments
4.2. Antiproliferation Assays
4.2.1. Monolayers
4.2.2. Spheroids
4.3. RNA Analyses
4.4. Immunoblotting
4.5. Preclinical Experiments
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef]
- Marsh, S.; McLeod, H.; Dolan, E.; Shukla, S.J.; Rabik, C.A.; Gong, L.; Hernandez-Boussard, T.; Lou, X.J.; Klein, T.E.; Altman, R.B. Platinum pathway. Pharm. Genom. 2009, 19, 563–564. [Google Scholar] [CrossRef] [Green Version]
- Evans, E.; Moggs, J.G.; Hwang, J.R.; Egly, J.M.; Wood, R.D. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997, 16, 6559–6573. [Google Scholar] [CrossRef] [Green Version]
- Volker, M.; Moné, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.; van Driel, R.; van Zeeland, A.A.; Mullenders, L.H. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nat. Cell Biol. 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Sameer, A.S.; Nissar, S. XPD—The lynchpin of NER: Molecule, Gene, Polymorphisms, and Role in Colorectal Carcinogenesis. Front. Mol. Biosci. 2018, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Kuper, J.; Kisker, C. Role of XPD in cellular functions: To TFIIH and beyond. DNA Repair 2016, 44, 136–142. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-Y.; Lee, H.-H.; Huang, C.-W.; Huang, C.-M.; Ma, C.-J.; Yin, T.-C.; Tsai, H.-L.; Chai, C.-Y.; Chen, Y.-T.; Wang, J.-Y. ERCC overexpression associated with a poor response of cT4b colorectal cancer with FOLFOX-based neoadjuvant concurrent chemoradiation. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, P.; Johnson, G.S.; Li, L.; Chen, Y.-S.; Dashwood, W.M.; Nguyen, N.; Ulusan, A.M.; Ertem, F.U.; Zhang, M.; Li, J.; et al. Acetylation of CCAR2 Establishes a BET/BRD9 acetyl switch in response to combined deacetylase and bromodomain inhibition. Cancer Res. 2019, 79, 918–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonkwo, A.; Mitra, J.; Johnson, G.S.; Li, L.; Dashwood, W.M.; Hegde, M.L.; Rajendran, P. Heterocyclic analogs of sulforaphane trigger DNA damage and impede DNA repair in colon cancer cells: Interplay of HATs and HDACs. Mol. Nutr. Food Res. 2018, 62, e1800228. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.-W.; Dashwood, W.-M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [Green Version]
- Wuerzberger-Davis, S.M.; Chang, P.-Y.; Berchtold, C.; Miyamoto, S. Enhanced G2-M Arrest by Nuclear Factor-κB-Dependent p21waf1/cip1 Induction. Mol. Cancer Res. 2005, 3, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Ho, E.; Williams, D.E.; Dashwood, R.H. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin. Epigenetics 2011, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Rassool, F.V. HDAC inhibitors: Roles of DNA damage and repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 1–13. [Google Scholar] [CrossRef]
- Chiu, L.-Y.; Gong, F.; Miller, K.M. Bromodomain proteins: Repairing DNA damage within chromatin. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160286. [Google Scholar] [CrossRef]
- López-Saavedra, A.; Gómez-Cabello, D.; Domínguez-Sánchez, M.S.; Mejías-Navarro, F.; Fernández-Ávila, M.J.; Dinant, C.; Martínez-Macías, M.I.; Bartek, C.D.J.; Huertas, P. A genome-wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat. Commun. 2016, 7, 12364. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.S.; Rajendran, P.; Dashwood, R.H. CCAR1 and CCAR2 as gene chameleons with antagonistic duality: Preclinical, human translational, and mechanistic basis. Cancer Sci. 2020, 111, 3416–3425. [Google Scholar] [CrossRef]
- Li, Q.; Damish, A.W.; Frazier, Z.; Liu, D.; Reznichenko, E.; Kamburov, A.; Bell, A.; Zhao, H.; Jordan, E.J.; Gao, S.P.; et al. ERCC2 helicase domain mutations confer nucleotide excision repair deficiency and drive cisplatin sensitivity in muscle-invasive bladder cancer. Clin. Cancer Res. 2019, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2D vs. 3D cell culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/ SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Damiani, E.; Duran, M.N.; Mohan, N.; Rajendran, P.; Dashwood, R.H. Targeting epigenetic ‘Readers’ with natural compounds for cancer interception. J. Cancer Prev. 2020, 25, 189–203. [Google Scholar] [CrossRef]
- Wang, R.; Dashwood, W.M.; Nian, H.; Löhr, C.V.; Fischer, K.A.; Tsuchiya, N.; Dashwood, R.H. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (PhIP). Int. J. Cancer 2011, 128, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, M.A.; Dashwood, W.M.; Wang, R.; Saeed, H.H.; Williams, D.E.; Ho, E.; Dashwood, R.H. A role for low-abundance miRNAs in colon cancer: The miR-206/ Krüppel-like factor 4 (KLF4) axis. Clin. Epigenet. 2012, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.; Ito, E.; Lau, K.S.; Mocanu, J.D.; Bastianutto, C.; Schimmer, A.D.; Liu, F.F. Increased efficiency for performing colony formation assays in 96-well plates: Novel applications to combination therapies and high-throughput screening. Biotechniques 2008, 44, ix–xiv. [Google Scholar] [CrossRef]
- Jaganathan, H.; Gage, J.A.; Leonard, F.; Srinivasan, S.; Souza, G.R.; Dave, B.; Godin, B. Three-dimensional In Vitro Co-Culture model of breast tumor using magnetic levitation. Sci. Rep. 2015, 4, 6468. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.; Gage, J.A.; Shen, T.; Haisler, W.L.; Neeley, S.K.; Shiao, S.; Chen, J.; Desai, P.K.; Liao, A.; Hebel, C.; et al. A spheroid toxicity assay using magnetic 3D bioprinting and real-time mobile device-based imaging. Sci. Rep. 2015, 5, 13987. [Google Scholar] [CrossRef] [Green Version]
- Ertem, F.U.; Zhang, W.; Chang, K.; Mohaiza Dashwood, W.; Rajendran, P.; Sun, D.; Dashwood, R.H. Oncogenic targets Mmp7, S100a9, Nppb and Aldh1a3 from transcriptome profiling of FAP and Pirc adenomas are downregulated in response to tumor suppression by Clotam. Int. J. Cancer 2017, 140, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Brzeszczyńska, J.; Brzeszczyński, F.; Samuel, K.; Morgan, K.; Morley, S.D.; Plevris, J.N.; Hayes, P.C. Validation of reference genes for gene expression studies by RT-qPCR in HepaRG cells during toxicity testing and disease modelling. Cells 2020, 9, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos-Landgraf, J.M.; Kwong, L.N.; Kendziorski, C.M.; Reichelderfer, M.; Torrealba, J.; Weichert, J.; Haag, J.D.; Chen, K.-S.; Waller, J.L.; Gould, M.N.; et al. A target-selected APC-mutant rat kindred enhances the modeling of familial human colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 4036–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulusan, A.M.; Rajendran, P.; Dashwood, W.M.; Yavuz, O.F.; Kapoor, S.; Gustafson, T.A.; Savage, M.I.; Brown, P.H.; Sei, S.; Mohammed, A.; et al. Optimization of erlotinib plus sulindac dosing regimens for intestinal cancer prevention in an APC-mutant model of Familial Adenomatous Polyposis (FAP). Cancer Prev. Res. 2021, 14, 325–336. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, S.; Gustafson, T.; Zhang, M.; Chen, Y.-S.; Li, J.; Nguyen, N.; Perez, J.E.T.; Dashwood, W.M.; Rajendran, P.; Dashwood, R.H. Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells. Cancers 2021, 13, 1438. https://doi.org/10.3390/cancers13061438
Kapoor S, Gustafson T, Zhang M, Chen Y-S, Li J, Nguyen N, Perez JET, Dashwood WM, Rajendran P, Dashwood RH. Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells. Cancers. 2021; 13(6):1438. https://doi.org/10.3390/cancers13061438
Chicago/Turabian StyleKapoor, Sabeeta, Trace Gustafson, Mutian Zhang, Ying-Shiuan Chen, Jia Li, Nhung Nguyen, Jorge Enrique Tovar Perez, Wan Mohaiza Dashwood, Praveen Rajendran, and Roderick H. Dashwood. 2021. "Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells" Cancers 13, no. 6: 1438. https://doi.org/10.3390/cancers13061438
APA StyleKapoor, S., Gustafson, T., Zhang, M., Chen, Y. -S., Li, J., Nguyen, N., Perez, J. E. T., Dashwood, W. M., Rajendran, P., & Dashwood, R. H. (2021). Deacetylase Plus Bromodomain Inhibition Downregulates ERCC2 and Suppresses the Growth of Metastatic Colon Cancer Cells. Cancers, 13(6), 1438. https://doi.org/10.3390/cancers13061438