
Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis


 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
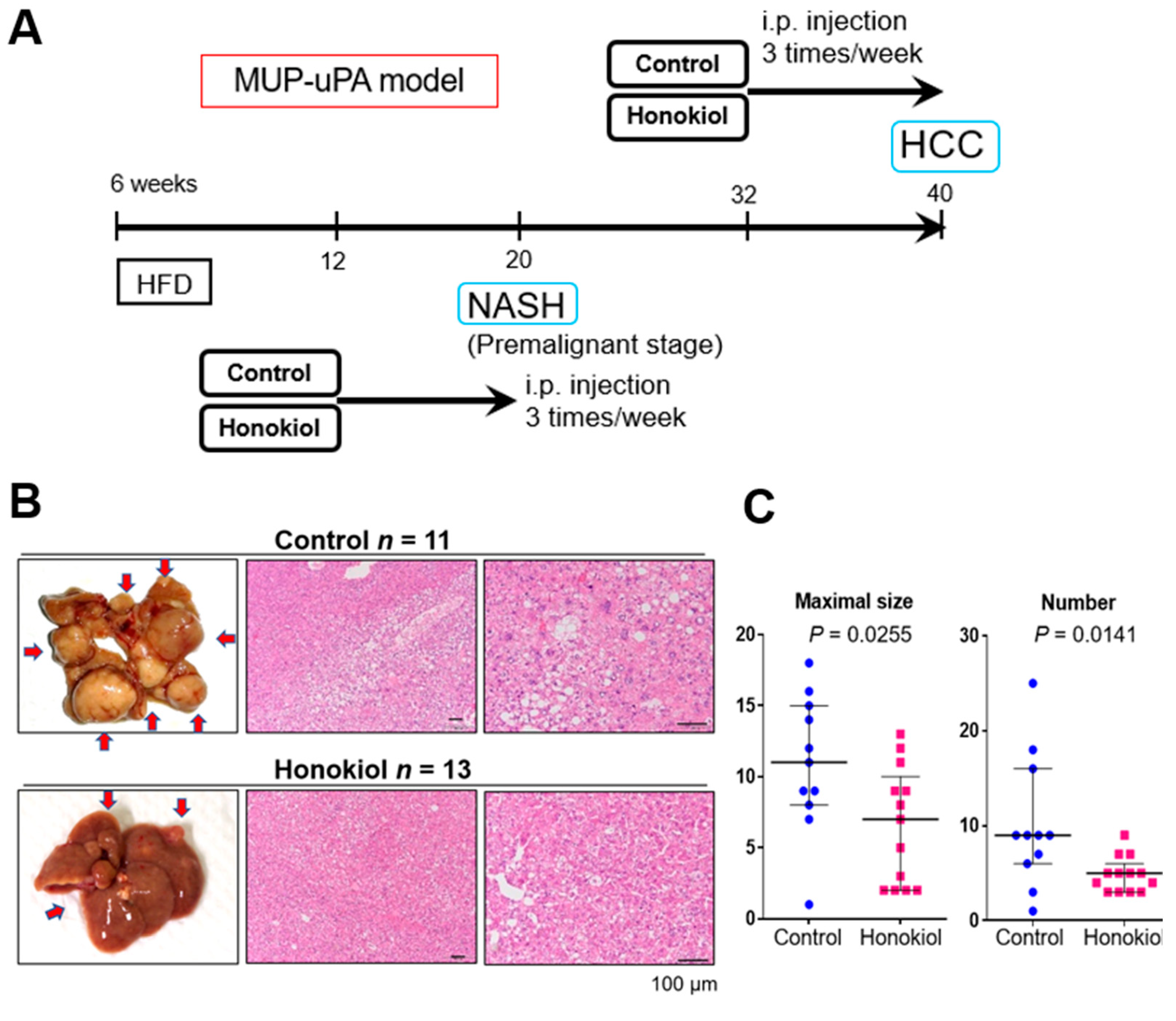
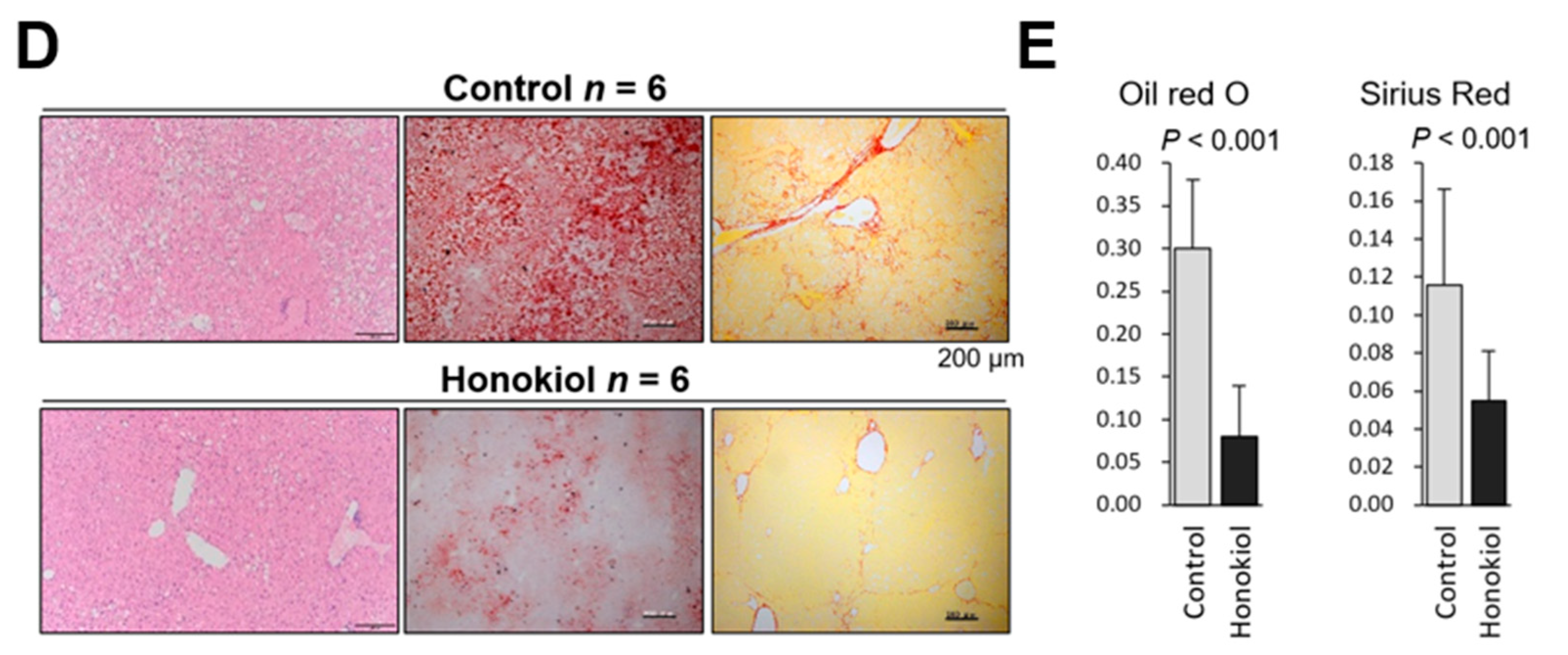
2.1. HNK Treatment Attenuates HCC Development in MUP-uPA Mice
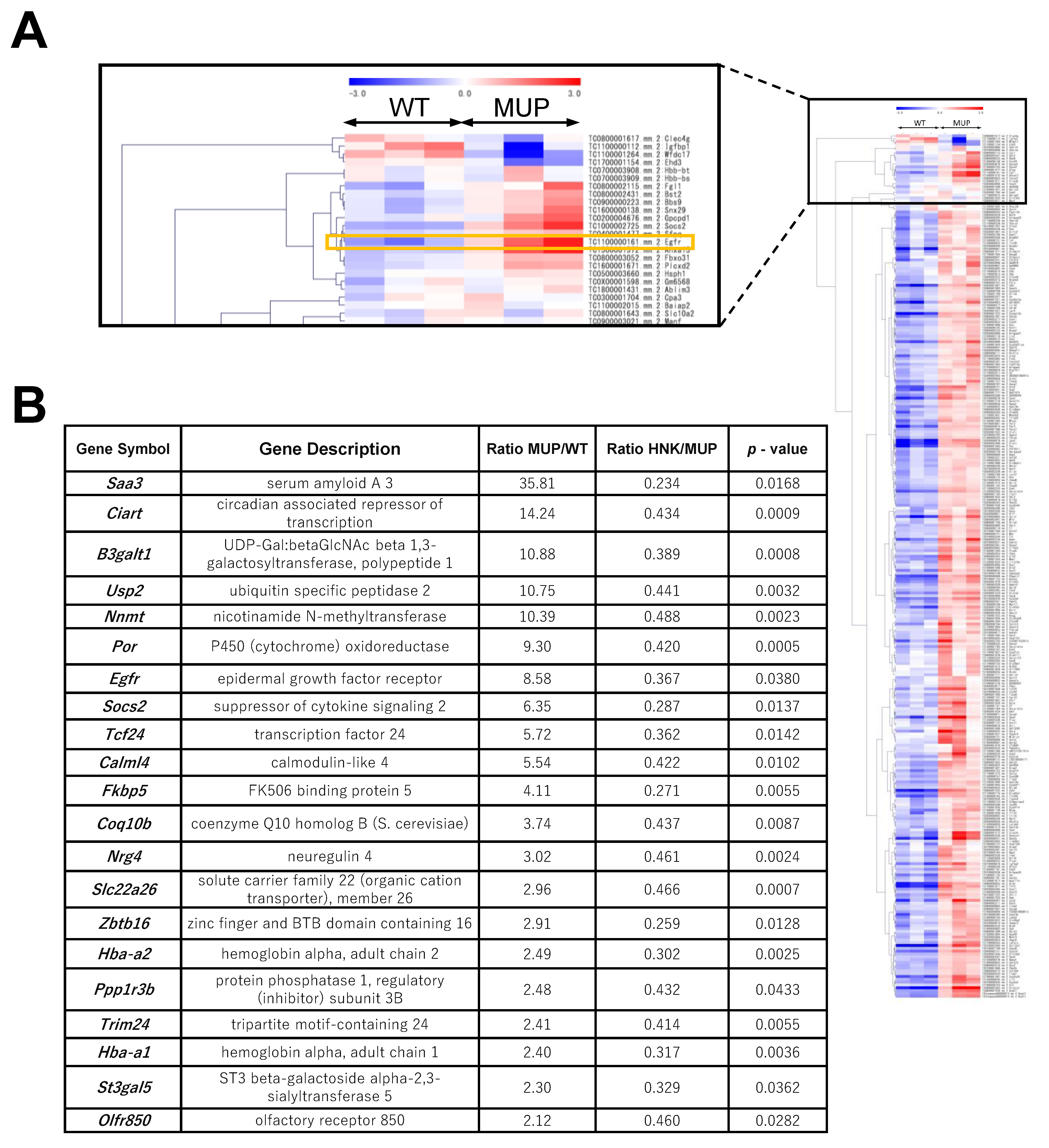
2.2. EGFR Signaling Is Upregulated in the HFD-fed MUP-uPA Mouse Liver
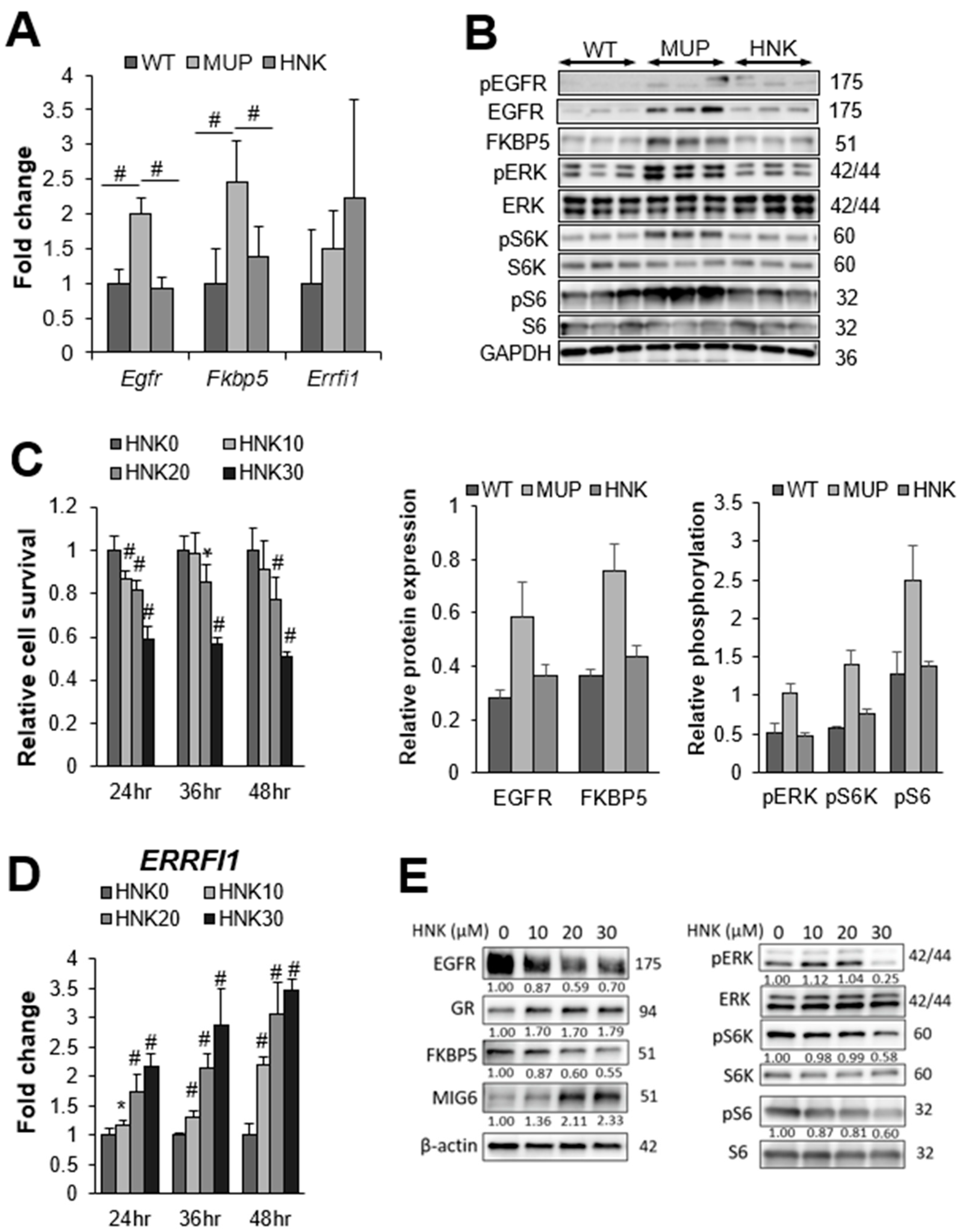
2.3. HNK Treatment Suppresses EGFR Signaling in the NASH Liver
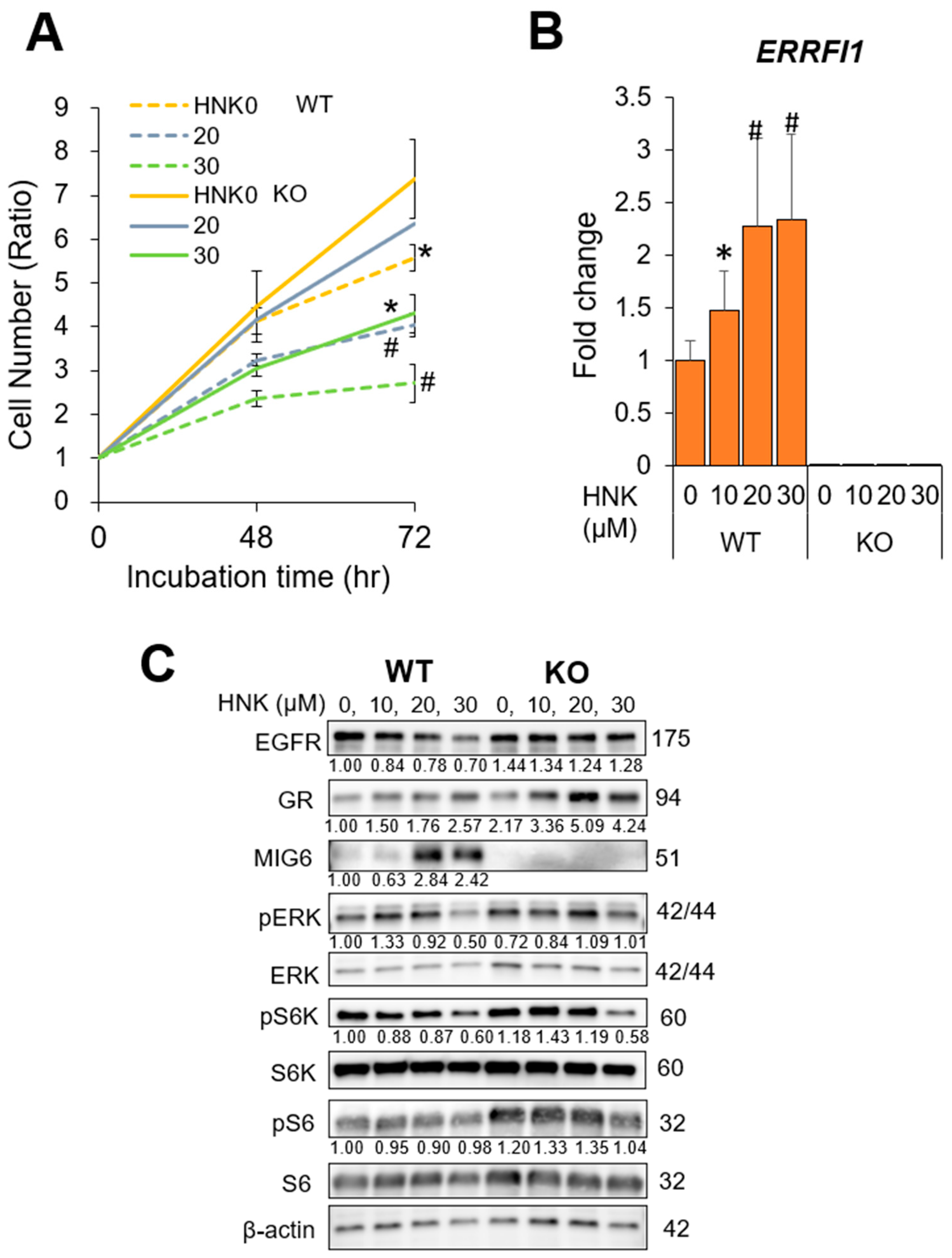
2.4. HNK Induces MIG6 Expression Leading to EGFR Downregulation
2.5. MIG6 Knockout (KO) Abrogates the Inhibitory Effects of HNK on HCC Cell Proliferation
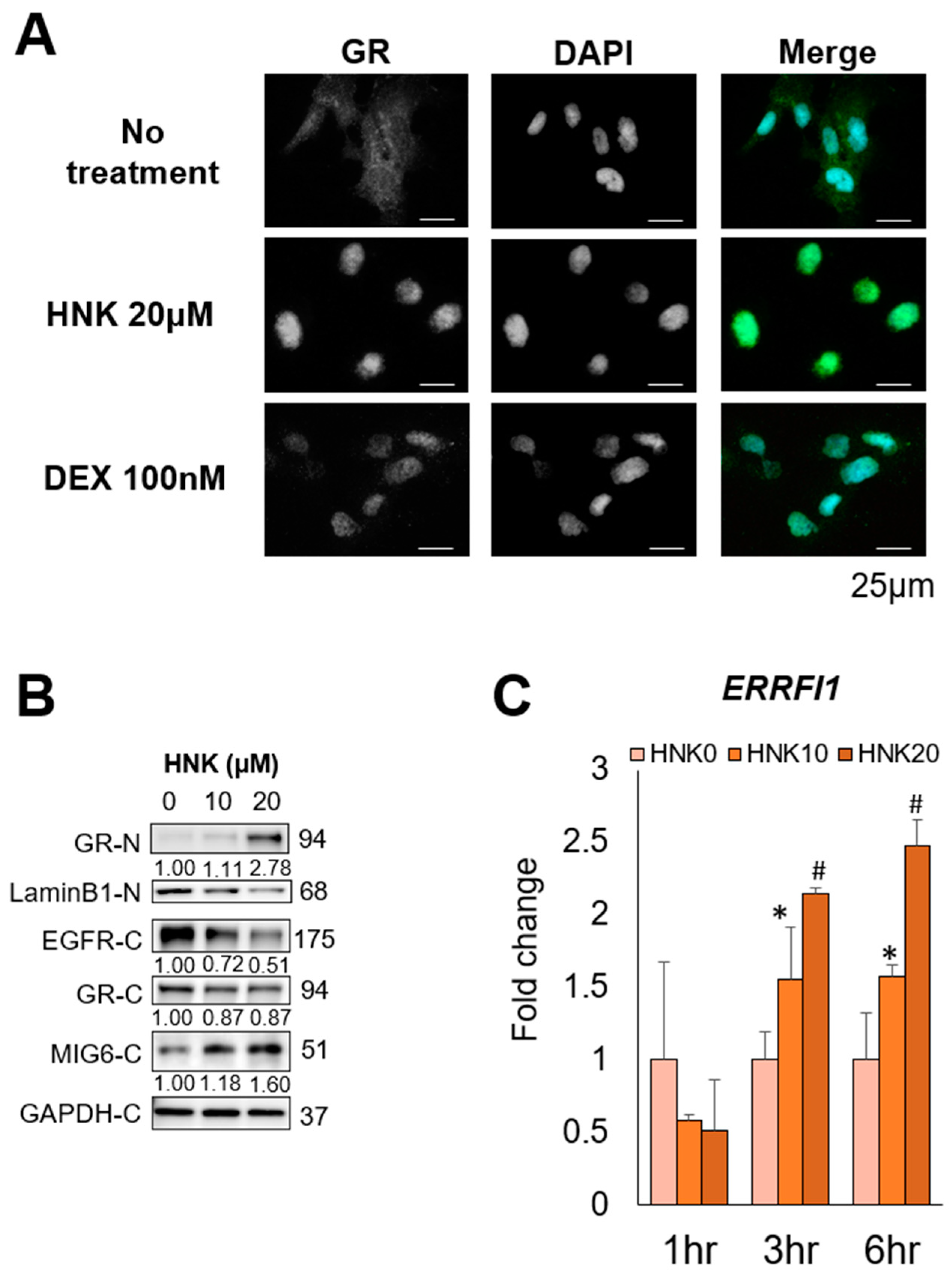
2.6. HNK Induces GR Nuclear Translocation, Leading to MIG6 Induction
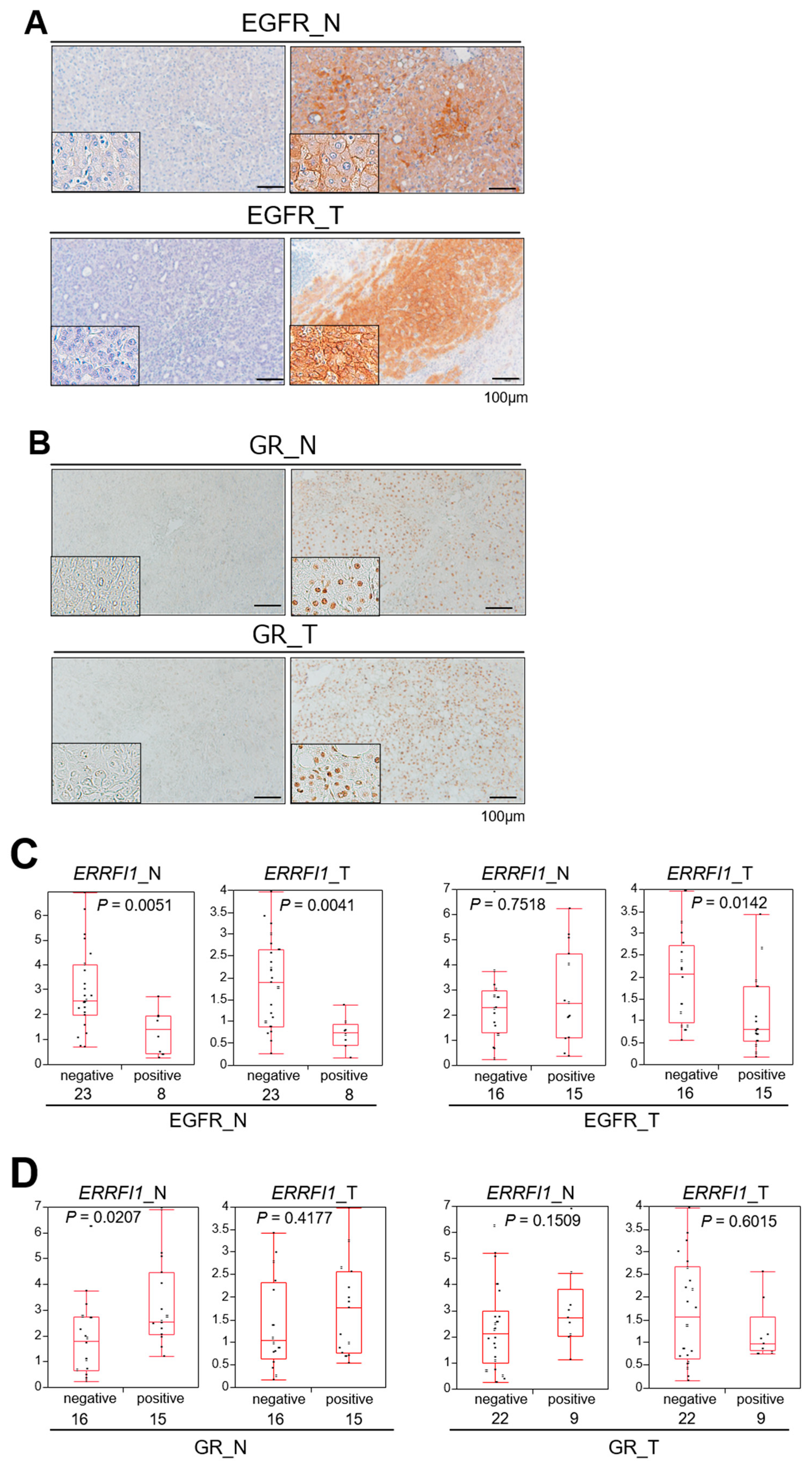
2.7. GR Activation and ERRFI1 Expression Are Inversely Correlated with EGFR Expression in Human HCC
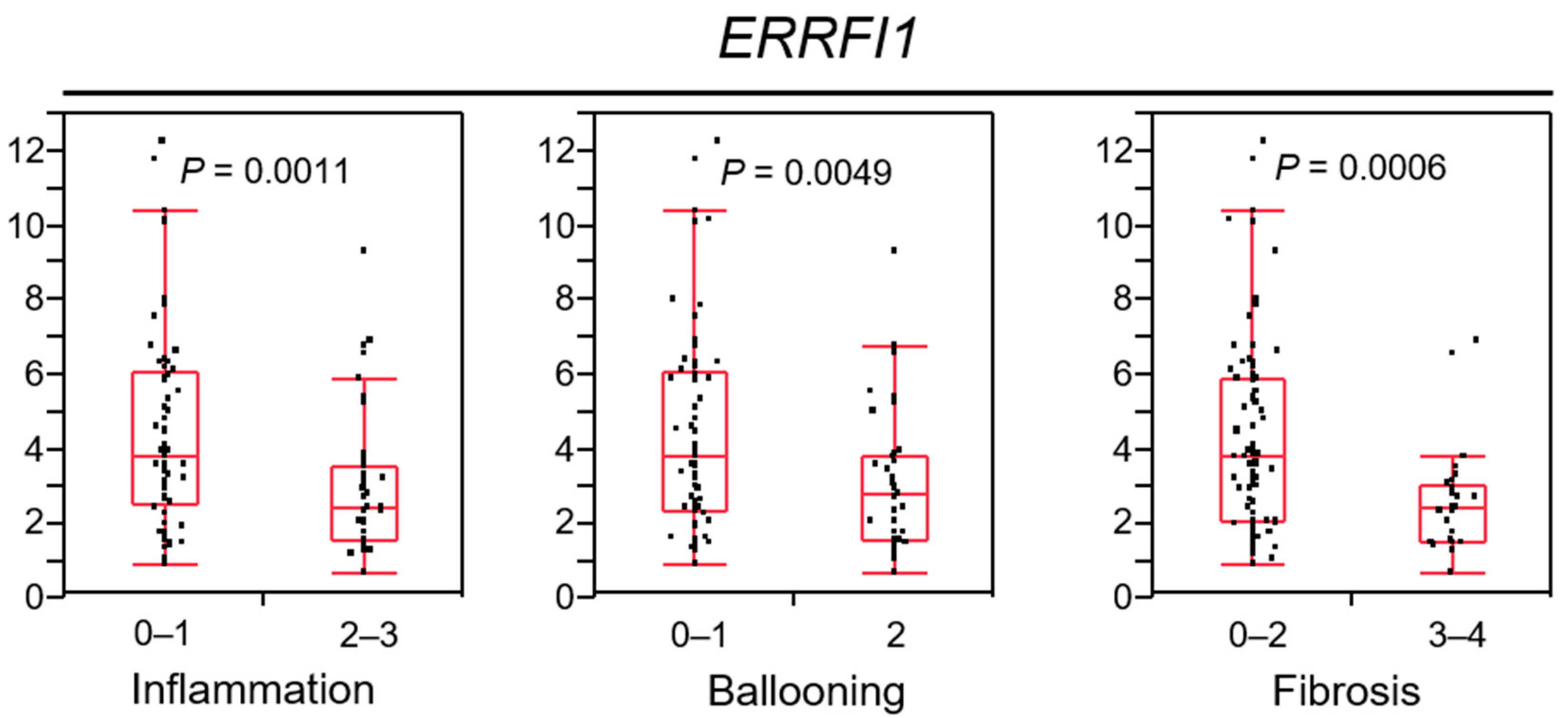
2.8. ERRFI1 Expression Decreases in Parallel with NAFLD/NASH Progression
2.9. HNK Treatment also Attenuates a NAFLD-HCC Model
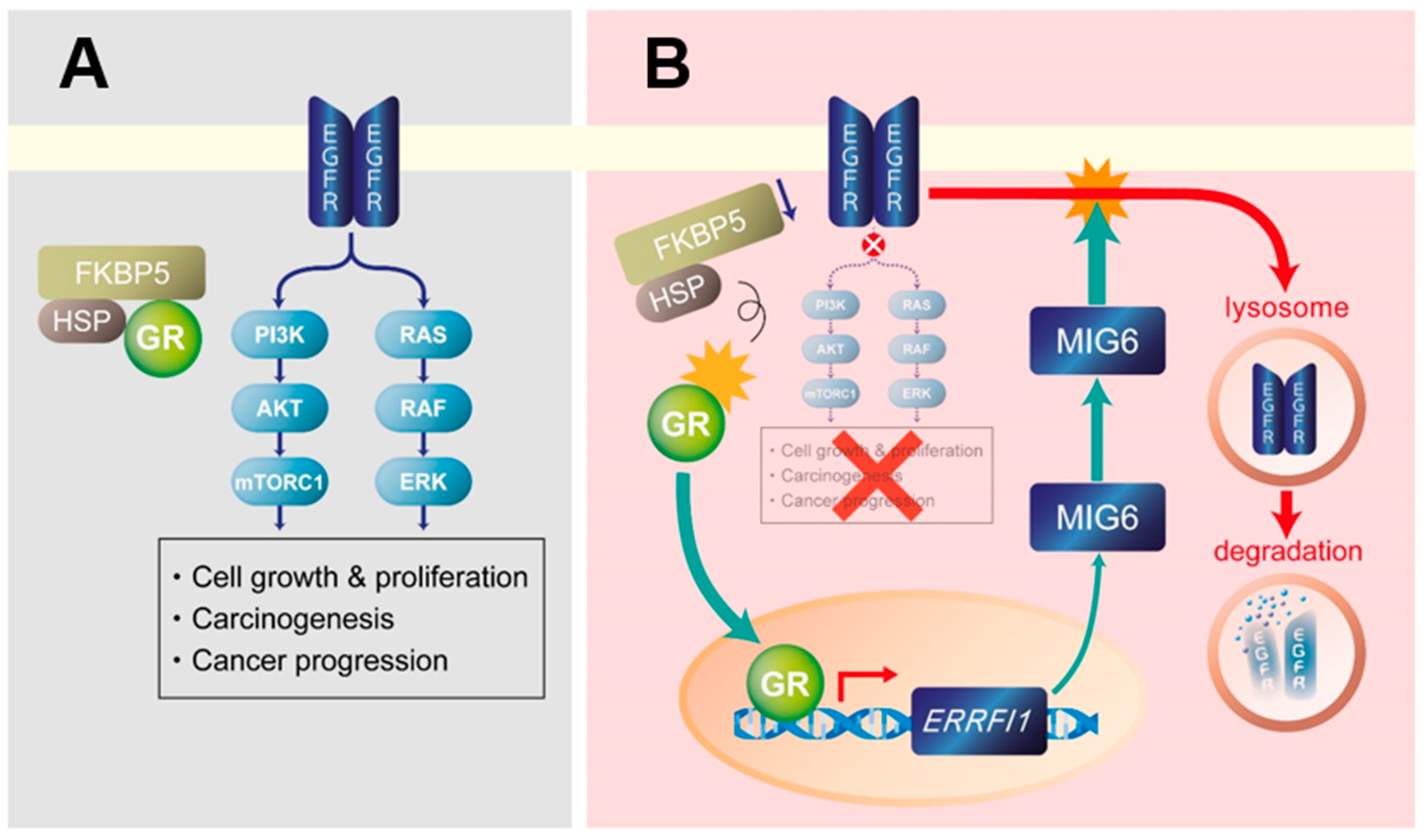
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Human Samples
4.3. Liver Histology
4.4. RNA Array Analysis
4.5. RNA Isolation and Quantitative RT-PCR
4.6. Human HCC Cells
4.7. Immunoblot Analysis
4.8. Immunofluorescence Analysis
4.9. Clonal Line Generation of MIG6/ERBB Receptor Feedback Inhibitor 1 (ERRFI1)-Knockout (KO) HCC Cells
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. SEARCH: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef]
- Paranjpe, S.; Bowen, W.C.; Mars, W.M.; Orr, A.; Haynes, M.M.; DeFrances, M.C.; Liu, S.; Tseng, G.C.; Tsagianni, A.; Michalopoulos, G.K. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016, 64, 1711–1724. [Google Scholar] [CrossRef]
- Bhushan, B.; Banerjee, S.; Paranjpe, S.; Koral, K.; Mars, W.M.; Stoops, J.W.; Orr, A.; Bowen, W.C.; Locker, J.; Michalopoulos, G.K. Pharmacologic Inhibition of Epidermal Growth Factor Receptor Suppresses Nonalcoholic Fatty Liver Disease in a Murine Fast-Food Diet Model. Hepatology 2019, 70, 1546–1563. [Google Scholar] [CrossRef]
- Choung, S.; Kim, J.M.; Joung, K.H.; Lee, E.S.; Kim, H.J.; Ku, B.J. Epidermal growth factor receptor inhibition attenuates non-alcoholic fatty liver disease in diet-induced obese mice. PLoS ONE 2019, 14, e0210828. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Yang, J.; Pei, H.; Luo, H.; Fu, A.; Yang, H.; Hu, J.; Zhao, C.; Chai, L.; Chen, X.; Shao, X.; et al. Non-toxic dose of liposomal honokiol suppresses metastasis of hepatocellular carcinoma through destabilizing EGFR and inhibiting the downstream pathways. Oncotarget 2017, 8, 915–932. [Google Scholar] [CrossRef]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E9879–E9888. [Google Scholar] [CrossRef]
- Li, L.; Lou, Z.; Wang, L. The role of FKBP5 in cancer aetiology and chemoresistance. Br. J. Cancer 2011, 104, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.J.; Diaz-Montero, C.M.; Mao, L.; Youssef, E.M.; Estecio, M.; Shen, L.; Issa, J.P. Epigenetic inactivation of EGFR by CpG island hypermethylation in cancer. Cancer Biol. Ther. 2006, 5, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Er, E.E.; Mendoza, M.C.; Mackey, A.M.; Rameh, L.E.; Blenis, J. AKT facilitates EGFR trafficking and degradation by phosphorylating and activating PIKfyve. Sci. Signal. 2013, 6, ra45. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, W.; Wen, J.; Ye, H.; Luo, H.; Bai, P.; Tang, M.; Wang, F.; Zheng, L.; Yang, S.; et al. Liposomal honokiol induced lysosomal degradation of Hsp90 client proteins and protective autophagy in both gefitinib-sensitive and gefitinib-resistant NSCLC cells. Biomaterials 2017, 141, 188–198. [Google Scholar] [CrossRef]
- Frosi, Y.; Anastasi, S.; Ballaro, C.; Varsano, G.; Castellani, L.; Maspero, E.; Polo, S.; Alema, S.; Segatto, O. A two-tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J. Cell Biol. 2010, 189, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Ferby, I.; Reschke, M.; Kudlacek, O.; Knyazev, P.; Pante, G.; Amann, K.; Sommergruber, W.; Kraut, N.; Ullrich, A.; Fassler, R.; et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat. Med. 2006, 12, 568–573. [Google Scholar] [CrossRef]
- Reschke, M.; Ferby, I.; Stepniak, E.; Seitzer, N.; Horst, D.; Wagner, E.F.; Ullrich, A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology 2010, 51, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Lauriola, M.; Enuka, Y.; Zeisel, A.; D’Uva, G.; Roth, L.; Sharon-Sevilla, M.; Lindzen, M.; Sharma, K.; Nevo, N.; Feldman, M.; et al. Diurnal suppression of EGFR signalling by glucocorticoids and implications for tumour progression and treatment. Nat. Commun. 2014, 5, 5073. [Google Scholar] [CrossRef]
- Sinclair, D.; Fillman, S.G.; Webster, M.J.; Weickert, C.S. Dysregulation of glucocorticoid receptor co-factors FKBP5, BAG1 and PTGES3 in prefrontal cortex in psychotic illness. Sci. Rep. 2013, 3, 3539. [Google Scholar] [CrossRef]
- Hapgood, J.P.; Avenant, C.; Moliki, J.M. Glucocorticoid-independent modulation of GR activity: Implications for immunotherapy. Pharmacol. Ther. 2016, 165, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Umemura, A.; Park, E.J.; Taniguchi, K.; Lee, J.H.; Shalapour, S.; Valasek, M.A.; Aghajan, M.; Nakagawa, H.; Seki, E.; Hall, M.N.; et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014, 20, 133–144. [Google Scholar] [CrossRef]
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- El-Bassiouni, A.; Nosseir, M.; Zoheiry, M.; El-Ahwany, E.; Ghali, A.; El-Bassiouni, N. Immunohistochemical expression of CD95 (Fas), c-myc and epidermal growth factor receptor in hepatitis C virus infection, cirrhotic liver disease and hepatocellular carcinoma. APMIS J. Pathol. Microbiol. Immunol. 2006, 114, 420–427. [Google Scholar] [CrossRef]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. AJCP Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef]
- Yoneda, N.; Sato, Y.; Kitao, A.; Ikeda, H.; Sawada-Kitamura, S.; Miyakoshi, M.; Harada, K.; Sasaki, M.; Matsui, O.; Nakanuma, Y. Epidermal growth factor induces cytokeratin 19 expression accompanied by increased growth abilities in human hepatocellular carcinoma. Lab. Investig. 2011, 91, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Antonucci, L.; Nakagawa, H.; Kim, J.Y.; Glitzner, E.; Caruso, S.; Shalapour, S.; Yang, L.; Valasek, M.A.; Lee, S.; et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 2018, 33, 1061–1077.e6. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol 2005, 23, 6657–6663. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Staal, B.; Su, Y.; Swiatek, P.; Zhao, P.; Cao, B.; Resau, J.; Sigler, R.; Bronson, R.; Vande Woude, G.F. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene 2007, 26, 269–276. [Google Scholar] [CrossRef]
- Amatschek, S.; Koenig, U.; Auer, H.; Steinlein, P.; Pacher, M.; Gruenfelder, A.; Dekan, G.; Vogl, S.; Kubista, E.; Heider, K.-H.; et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res. 2004, 64, 844–856. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [CrossRef]
- Li, Z.; Qu, L.; Luo, W.; Tian, Y.; Zhai, H.; Xu, K.; Zhong, H. Mig-6 is down-regulated in HCC and inhibits the proliferation of HCC cells via the P-ERK/Cyclin D1 pathway. Exp. Mol. Pathol. 2017, 102, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, Y.; He, H.T.; Yang, Q. MiR-589-5p is a potential prognostic marker of hepatocellular carcinoma and regulates tumor cell growth by targeting MIG-6. Neoplasma 2018, 65, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, H.; Wang, H.; Dong, Y.; Yin, M.; Zhang, L.; Wei, J. MicroRNA-374a Promotes Hepatocellular Carcinoma Cell Proliferation by Targeting Mitogen-Inducible Gene 6 (MIG-6). Oncol. Res. 2018, 26, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef] [PubMed]
- Balan, M.; Chakraborty, S.; Flynn, E.; Zurakowski, D.; Pal, S. Honokiol inhibits c-Met-HO-1 tumor-promoting pathway and its cross-talk with calcineurin inhibitor-mediated renal cancer growth. Sci. Rep. 2017, 7, 5900. [Google Scholar] [CrossRef]
- Singh, T.; Katiyar, S.K. Honokiol inhibits non-small cell lung cancer cell migration by targeting PGE2-mediated activation of β-catenin signaling. PLoS ONE 2013, 8, e60749. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Bhardwaj, A.; Srivastava, S.K.; Singh, S.; McClellan, S.; Wang, B.; Singh, A.P. Honokiol arrests cell cycle, induces apoptosis, and potentiates the cytotoxic effect of gemcitabine in human pancreatic cancer cells. PLoS ONE 2011, 6, e21573. [Google Scholar] [CrossRef] [PubMed]
- Maioli, M.; Basoli, V.; Carta, P.; Fabbri, D.; Dettori, M.A.; Cruciani, S.; Serra, P.A.; Delogu, G. Synthesis of magnolol and honokiol derivatives and their effect against hepatocarcinoma cells. PLoS ONE 2018, 13, e0192178. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef]
- Bai, X.; Cerimele, F.; Ushio-Fukai, M.; Waqas, M.; Campbell, P.M.; Govindarajan, B.; Der, C.J.; Battle, T.; Frank, D.A.; Ye, K.; et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003, 278, 35501–35507. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2017, 54, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, J.; Wang, X.; Zhao, X.; Li, Z.; Niu, J.; Steer, C.J.; Zheng, G.; Song, G. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kappaB-TNFalpha pathway. J. Hepatol 2019, 70, 87–96. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuda, K.; Umemura, A.; Umemura, S.; Kataoka, S.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; Moriguchi, M.; Kanbara, Y.; et al. Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis. Cancers 2021, 13, 1515. https://doi.org/10.3390/cancers13071515
Okuda K, Umemura A, Umemura S, Kataoka S, Taketani H, Seko Y, Nishikawa T, Yamaguchi K, Moriguchi M, Kanbara Y, et al. Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis. Cancers. 2021; 13(7):1515. https://doi.org/10.3390/cancers13071515
Chicago/Turabian StyleOkuda, Keiichiro, Atsushi Umemura, Shiori Umemura, Seita Kataoka, Hiroyoshi Taketani, Yuya Seko, Taichiro Nishikawa, Kanji Yamaguchi, Michihisa Moriguchi, Yoshihiro Kanbara, and et al. 2021. "Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis" Cancers 13, no. 7: 1515. https://doi.org/10.3390/cancers13071515
APA StyleOkuda, K., Umemura, A., Umemura, S., Kataoka, S., Taketani, H., Seko, Y., Nishikawa, T., Yamaguchi, K., Moriguchi, M., Kanbara, Y., Arbiser, J. L., Shima, T., Okanoue, T., Karin, M., & Itoh, Y. (2021). Honokiol Prevents Non-Alcoholic Steatohepatitis-Induced Liver Cancer via EGFR Degradation through the Glucocorticoid Receptor—MIG6 Axis. Cancers, 13(7), 1515. https://doi.org/10.3390/cancers13071515