
An Fc-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia

 ,
, 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibody Production and Purification
2.2. Cell Lines and Primary Material
2.3. Flow Cytometry
2.4. Analysis of NK Cell Activation and Degranulation
2.5. Analysis of NK Cell Cytotoxicity
2.6. Statistics
3. Results
3.1. Characterization of Fc-Optimized 293C3-SDIE on B-ALL Cells
3.2. Induction of NK Cell Reactivity against B-ALL Cell Lines
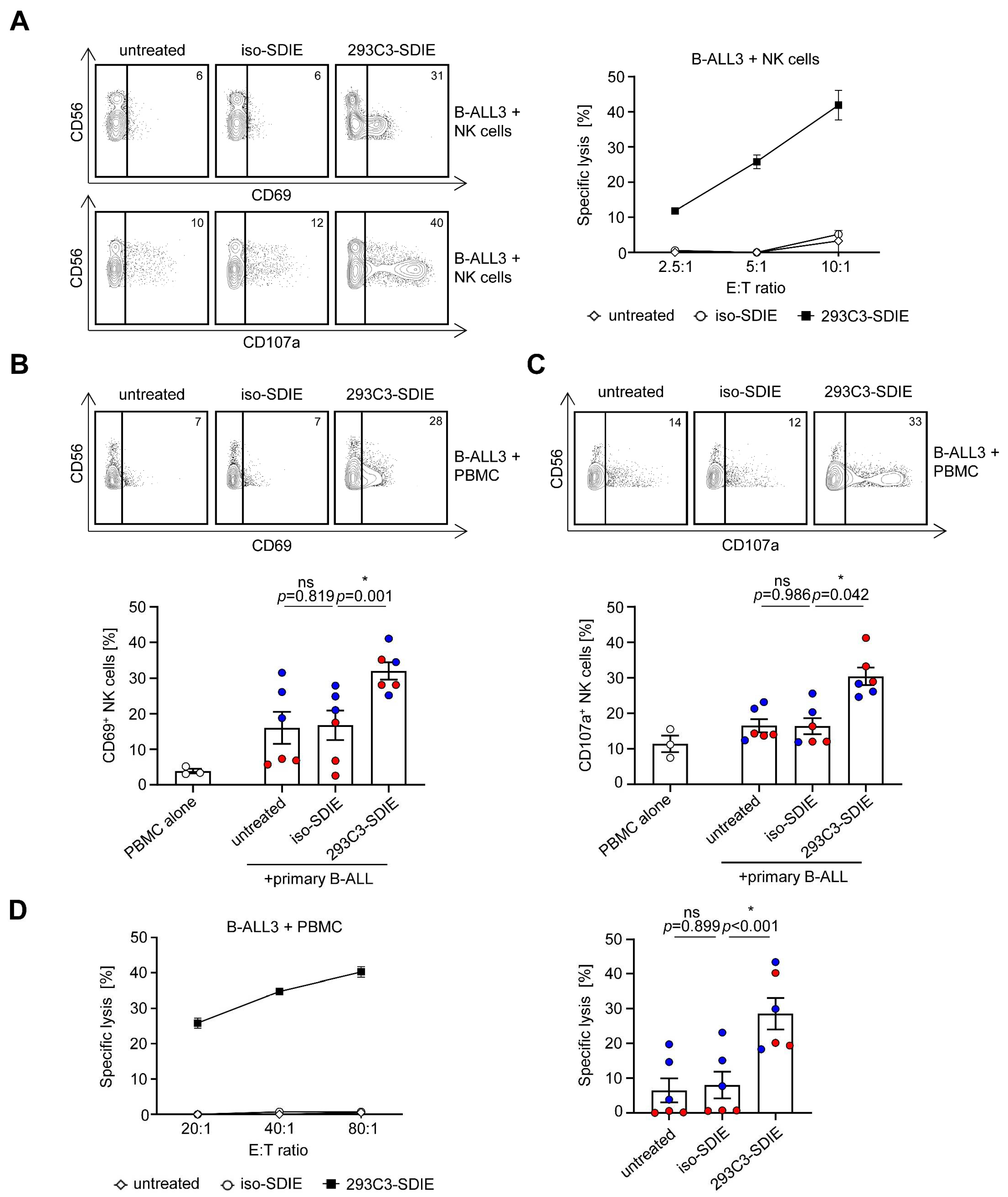
3.3. Induction of NK Cell Reactivity against Primary B-ALL Cells
3.4. Clinical Characteristics of Primary B-ALL Patients and Prognostic Evaluation of CD133 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keating, G.M. Rituximab: A review of its use in chronic lymphocytic leukaemia, low-grade or follicular lymphoma and diffuse large B-cell lymphoma. Drugs 2010, 70, 1445–1476. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [Green Version]
- Schmied, B.J.; Lutz, M.S.; Riegg, F.; Zekri, L.; Heitmann, J.S.; Buhring, H.J.; Jung, G.; Salih, H.R. Induction of NK Cell Reactivity against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody. Cancers 2019, 11, 1966. [Google Scholar] [CrossRef] [Green Version]
- Van der Horst, H.J.; Nijhof, I.S.; Mutis, T.; Chamuleau, M.E.D. Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers 2020, 12, 3041. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.S.; Illmer, T.; et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Scheible, C.A.; Nuebling, T.; Kopp, H.G.; Wirths, S.; Azuma, M.; Schneider, P.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. RANKL expression, function, and therapeutic targeting in multiple myeloma and chronic lymphocytic leukemia. Cancer Res. 2013, 73, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Steinbacher, J.; Baltz-Ghahremanpour, K.; Schmiedel, B.J.; Steinle, A.; Jung, G.; Kubler, A.; Andre, M.C.; Grosse-Hovest, L.; Salih, H.R. An Fc-optimized NKG2D-immunoglobulin G fusion protein for induction of natural killer cell reactivity against leukemia. Int. J. Cancer 2015, 136, 1073–1084. [Google Scholar] [CrossRef]
- Raab, S.; Steinbacher, J.; Schmiedel, B.J.; Kousis, P.C.; Steinle, A.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. Fc-optimized NKG2D-Fc constructs induce NK cell antibody-dependent cellular cytotoxicity against breast cancer cells independently of HER2/neu expression status. J. Immunol. 2014, 193, 4261–4272. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, B.J.; Werner, A.; Steinbacher, J.; Nuebling, T.; Buechele, C.; Grosse-Hovest, L.; Salih, H.R. Generation and preclinical characterization of a Fc-optimized GITR-Ig fusion protein for induction of NK cell reactivity against leukemia. Mol. Ther. 2013, 21, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.; Grosse-Hovest, L.; Nubling, T.; Pyz, E.; Bamberg, M.L.; Aulwurm, S.; Buhring, H.J.; Schwartz, K.; Haen, S.P.; Schilbach, K.; et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012, 26, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Koerner, S.P.; Andre, M.C.; Leibold, J.S.; Kousis, P.C.; Kubler, A.; Pal, M.; Haen, S.P.; Buhring, H.J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2017, 31, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Schmied, B.J.; Riegg, F.; Zekri, L.; Grosse-Hovest, L.; Buhring, H.J.; Jung, G.; Salih, H.R. An Fc-Optimized CD133 Antibody for Induction of Natural Killer Cell Reactivity against Colorectal Cancer. Cancers 2019, 11, 789. [Google Scholar] [CrossRef] [Green Version]
- Bühring, H.-J.; Seiffert, M.; Marxer, A.; Faul, B.W.C.; Kanz, L.; Brugger, W. AC133 Antigen Expression Is Not Restricted to Acute Myeloid Leukemia Blasts But Is Also Found on Acute Lymphoid Leukemia Blasts and on a Subset of CD34+ B-Cell Precursors. Blood 1999, 94, 832–833. [Google Scholar] [CrossRef]
- Tolba, F.M.; Foda, M.E.; Kamal, H.M.; Elshabrawy, D.A. Expression of CD133 in acute leukemia. Med. Oncol. 2013, 30, 527. [Google Scholar] [CrossRef]
- Godfrey, L.; Crump, N.T.; O’Byrne, S.; Lau, I.J.; Rice, S.; Harman, J.R.; Jackson, T.; Elliott, N.; Buck, G.; Connor, C.; et al. H3K79me2/3 controls enhancer-promoter interactions and activation of the pan-cancer stem cell marker PROM1/CD133 in MLL-AF4 leukemia cells. Leukemia 2021, 35, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Mak, A.B.; Nixon, A.M.; Moffat, J. The mixed lineage leukemia (MLL) fusion-associated gene AF4 promotes CD133 transcription. Cancer Res. 2012, 72, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Tae Lee, S.; Ho Jang, J.; Hong Min, Y.; Sook Hahn, J.; Woong Ko, Y. AC133 antigen as a prognostic factor in acute leukemia. Leuk. Res. 2001, 25, 757–767. [Google Scholar] [CrossRef]
- Wuchter, C.; Ratei, R.; Spahn, G.; Schoch, C.; Harbott, J.; Schnittger, S.; Haferlach, T.; Creutzig, U.; Sperling, C.; Karawajew, L.; et al. Impact of CD133 (AC133) and CD90 expression analysis for acute leukemia immunophenotyping. Haematologica 2001, 86, 154–161. [Google Scholar]
- Batlevi, C.L.; Matsuki, E.; Brentjens, R.J.; Younes, A. Novel immunotherapies in lymphoid malignancies. Nat. Rev. Clin. Oncol. 2016, 13, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Maury, S.; Chevret, S.; Thomas, X.; Heim, D.; Leguay, T.; Huguet, F.; Chevallier, P.; Hunault, M.; Boissel, N.; Escoffre-Barbe, M.; et al. Rituximab in B-Lineage Adult Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 1044–1053. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef]
- Kellner, C.; Peipp, M.; Gramatzki, M.; Schrappe, M.; Schewe, D.M. Perspectives of Fc engineered antibodies in CD19 targeting immunotherapies in pediatric B-cell precursor acute lymphoblastic leukemia. Oncoimmunology 2018, 7, e1448331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From preclinical development to success in B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 96–104. [Google Scholar] [CrossRef]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; Gonzalez Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; Andre, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Hefazi, M.; Litzow, M.R. Recent advances in the biology and treatment of B-cell acute lymphoblastic leukemia. Blood Lymphat. Cancer 2018, 8, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Hu, Y.; Jin, Z.; Zhai, Y.; Tan, Y.; Sun, Y.; Zhu, S.; Zhao, C.; Chen, B.; Zhu, J.; et al. TanCAR T cells targeting CD19 and CD133 efficiently eliminate MLL leukemic cells. Leukemia 2018, 32, 2012–2016. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, J.D.; Fink, K.L.; Landolfi, J.C.; Markert, J.; Piccioni, D.E.; Glantz, M.J.; Swanson, S.J.; Gringeri, A.; Yu, J. Immunological targeting of CD133 in recurrent glioblastoma: A multi-center phase I translational and clinical study of autologous CD133 dendritic cell immunotherapy. J. Clin. Oncol. 2017, 35, 2059. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number of Patients (%) (n = 28) |
|---|---|
| Sex | |
| Male | 16 (57) |
| Female | 12 (43) |
| Median age (years) | 40 (range 0–81) |
| B-ALL type | |
| Pro-B-ALL | 5 (18) |
| Common ALL | 18 (64) |
| Pre-B-ALL | 5 (18) |
| BCR-ABL status | |
| positive | 12 (43) |
| negative | 16 (57) |
| MLL-AF4 status | |
| positive | 3 (11) |
| negative | 6 (21) |
| NA | 19 (68) |
| Blood count | |
| WBC (G/L) | 84 (range 4.7–463) |
| Hb (g/dL) | 9.6 (range 4.8–13.6) |
| Plt (G/L) | 80 (range 11–280) |
| Risk stratification * | |
| SR | 6 (21) |
| HR | 9 (32) |
| VHR | 12 (43) |
| NA | 1 (4) |
| Complete remission π | 15 (54) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riegg, F.; Lutz, M.S.; Schmied, B.J.; Heitmann, J.S.; Queudeville, M.; Lang, P.; Jung, G.; Salih, H.R.; Märklin, M. An Fc-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1632. https://doi.org/10.3390/cancers13071632
Riegg F, Lutz MS, Schmied BJ, Heitmann JS, Queudeville M, Lang P, Jung G, Salih HR, Märklin M. An Fc-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia. Cancers. 2021; 13(7):1632. https://doi.org/10.3390/cancers13071632
Chicago/Turabian StyleRiegg, Fabian, Martina S. Lutz, Bastian J. Schmied, Jonas S. Heitmann, Manon Queudeville, Peter Lang, Gundram Jung, Helmut R. Salih, and Melanie Märklin. 2021. "An Fc-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia" Cancers 13, no. 7: 1632. https://doi.org/10.3390/cancers13071632
APA StyleRiegg, F., Lutz, M. S., Schmied, B. J., Heitmann, J. S., Queudeville, M., Lang, P., Jung, G., Salih, H. R., & Märklin, M. (2021). An Fc-Optimized CD133 Antibody for Induction of NK Cell Reactivity against B Cell Acute Lymphoblastic Leukemia. Cancers, 13(7), 1632. https://doi.org/10.3390/cancers13071632