
Chromatin Regulator SPEN/SHARP in X Inactivation and Disease

 ,
,
Abstract
:Simple Summary
Abstract
1. Long Non-Coding RNAs and Cancer
2. The Inactive X Chromosome Status in Cancer
3. Chromatin Modifiers That Act in XCI
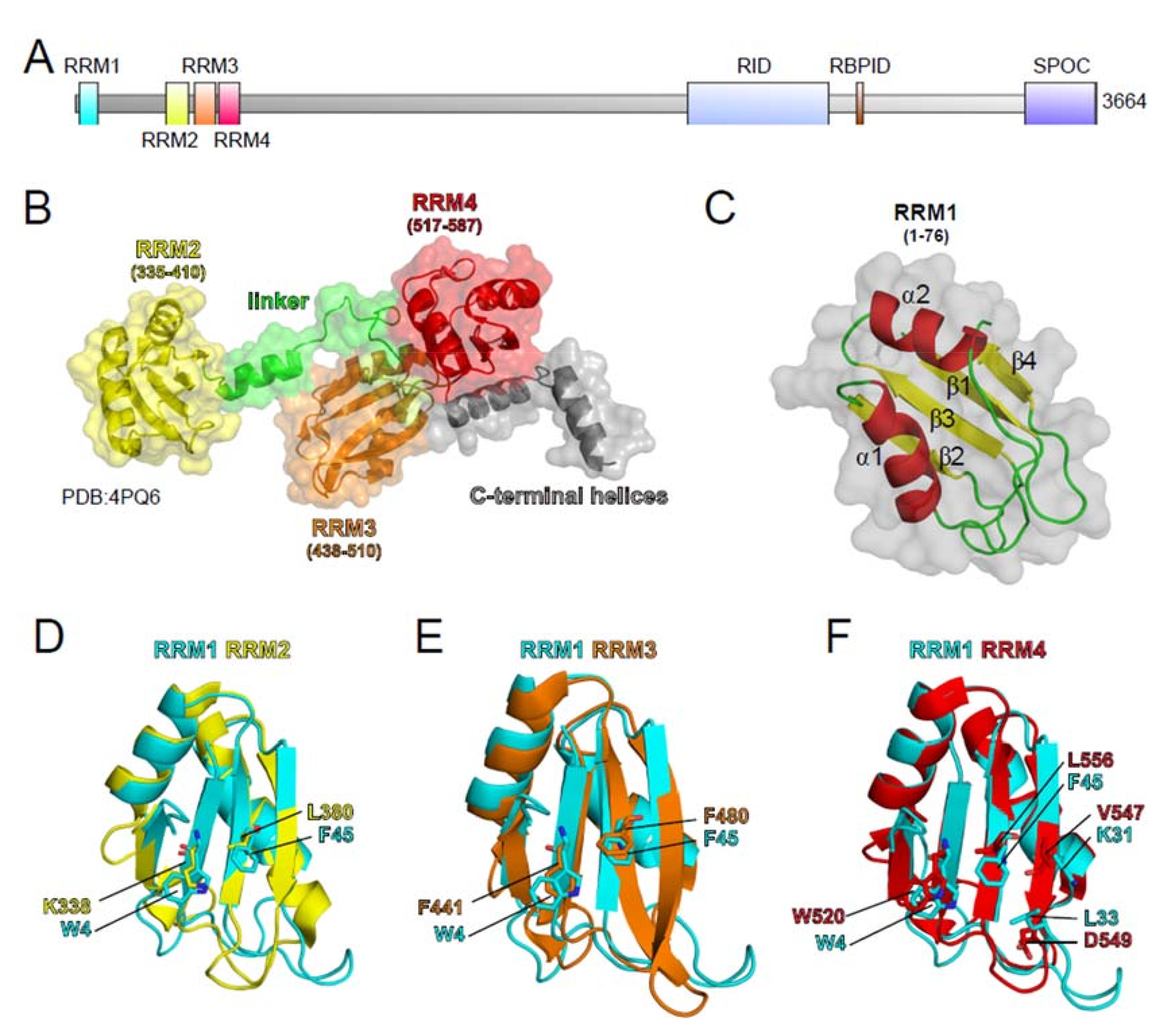
3.1. SHARP
3.1.1. SHARP in Chromatin Regulation
3.1.2. SHARP Directly Interacts with the NCoR/HDAC Complex
3.1.3. Pathological Deregulation of SHARP
3.2. PRC1 and PRC2
3.3. DNA Methyltransferases (DNMTs)
4. Conclusions
Funding
Conflicts of Interest
References
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Li, Y.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Thum, T.; Condorelli, G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ. Res. 2015, 116, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Liu, F.; Somarowthu, S.; Pyle, A.M. Visualizing the secondary and tertiary architectural domains of lncRNA RepA. Nat. Chem. Biol. 2017, 13, 282–289. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, Z.; Shi, H.; Li, H.; Li, L.; Fang, R.; Cai, X.; Liu, B.; Zhang, X.; Ye, L. HBXIP and LSD1 Scaffolded by lncRNA Hotair Mediate Transcriptional Activation by c-Myc. Cancer Res. 2016, 76, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Yang, F.Q.; Chen, S.J.; Che, J.; Zheng, J.H. Upregulation of long non-coding RNA MALAT1 correlates with tumor progression and poor prognosis in clear cell renal cell carcinoma. Tumour Biol. 2015, 36, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.X.; Wang, H.J.; Li, X.R.; Li, T.; Su, G.; Yang, P.; Wu, J.W. Long noncoding RNA MALAT1 associates with the malignant status and poor prognosis in glioma. Tumour Biol. 2015, 36, 3355–3359. [Google Scholar] [CrossRef]
- Dong, Y.; Liang, G.; Yuan, B.; Yang, C.; Gao, R.; Zhou, X. MALAT1 promotes the proliferation and metastasis of osteosarcoma cells by activating the PI3K/Akt pathway. Tumour Biol. 2015, 36, 1477–1486. [Google Scholar] [CrossRef]
- Cho, S.F.; Chang, Y.C.; Chang, C.S.; Lin, S.F.; Liu, Y.C.; Hsiao, H.H.; Chang, J.G.; Liu, T.C. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer 2014, 14, 809. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.T.; Shi, D.B.; Wang, Y.W.; Li, X.X.; Xu, Y.; Tripathi, P.; Gu, W.L.; Cai, G.X.; Cai, S.J. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3174–3181. [Google Scholar]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.S.; Wang, X.A.; Wu, W.G.; Hu, Y.P.; Li, M.L.; Ding, Q.; Weng, H.; Shu, Y.J.; Liu, T.Y.; Jiang, L.; et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 2014, 15, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Su, L.; Chen, X.; Li, P.; Cai, Q.; Yu, B.; Liu, B.; Wu, W.; Zhu, Z. MALAT1 promotes cell proliferation in gastric cancer by recruiting SF2/ASF. Biomed. Pharmacother. 2014, 68, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, D.; Li, Y.; Pan, X.; Zhang, X.; Kuang, B.; Zhou, M.; Li, X.; Xiong, W.; Li, G.; et al. The Long Noncoding RNA MALAT-1 is A Novel Biomarker in Various Cancers: A Meta-analysis Based on the GEO Database and Literature. J. Cancer 2016, 7, 991–1001. [Google Scholar] [CrossRef]
- Poirier, F.; Chan, C.T.; Timmons, P.M.; Robertson, E.J.; Evans, M.J.; Rigby, P.W. The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development 1991, 113, 1105–1114. [Google Scholar] [PubMed]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. Bioessays 2010, 32, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Ripoche, M.A.; Yoshimizu, T.; Dandolo, L. The H19 gene: Regulation and function of a non-coding RNA. Cytogenet. Genome Res. 2006, 113, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, B.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 2014, 5, 2318–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Bi, J.; Xue, X.; Zheng, L.; Zhi, K.; Hua, J.; Fang, G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012, 279, 3159–3165. [Google Scholar] [CrossRef]
- Yoshimura, H.; Matsuda, Y.; Yamamoto, M.; Kamiya, S.; Ishiwata, T. Expression and role of long non-coding RNA H19 in carcinogenesis. Front. Biosci. (Landmark Ed.) 2018, 23, 614–625. [Google Scholar] [CrossRef]
- Xie, S.S.; Jin, J.; Xu, X.; Zhuo, W.; Zhou, T.H. Emerging roles of non-coding RNAs in gastric cancer: Pathogenesis and clinical implications. World J. Gastroenterol. 2016, 22, 1213–1223. [Google Scholar] [CrossRef]
- Zhang, E.B.; Kong, R.; Yin, D.D.; You, L.H.; Sun, M.; Han, L.; Xu, T.P.; Xia, R.; Yang, J.S.; De, W.; et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 2014, 5, 2276–2292. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Xue, X.; Zheng, L.; Bi, J.; Zhou, Y.; Zhi, K.; Gu, Y.; Fang, G. Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. FEBS J. 2014, 281, 802–813. [Google Scholar] [CrossRef]
- Kong, R.; Zhang, E.B.; Yin, D.D.; You, L.H.; Xu, T.P.; Chen, W.M.; Xia, R.; Wan, L.; Sun, M.; Wang, Z.X.; et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol. Cancer 2015, 14, 82. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Wu, G.; Fan, H.; Wu, J.; Feng, J. Long noncoding RNA SPRY4-IT1 predicts poor patient prognosis and promotes tumorigenesis in gastric cancer. Tumour. Biol. 2015, 36, 6751–6758. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.Y.; Yu, Q.M.; Du, Y.A.; Yang, L.T.; Dong, R.Z.; Huang, L.; Yu, P.F.; Cheng, X.D. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 2013, 9, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Jutooru, I.; Chadalapaka, G.; Johnson, G.; Frank, J.; Burghardt, R.; Kim, S.; Safe, S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2013, 32, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Okamoto, K.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okada, Y. The roles of supernumerical X chromosomes and XIST expression in testicular germ cell tumors. J. Urol 2003, 169, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Silver, D.P.; Greenberg, R.A.; Avni, D.; Drapkin, R.; Miron, A.; Mok, S.C.; Randrianarison, V.; Brodie, S.; Salstrom, J.; et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell 2002, 111, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Pageau, G.J.; Hall, L.L.; Lawrence, J.B. BRCA1 does not paint the inactive X to localize XIST RNA but may contribute to broad changes in cancer that impact XIST and Xi heterochromatin. J. Cell Biochem. 2007, 100, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.L.; Wang, Z.C.; de Nicolo, A.; Lu, X.; Brown, M.; Miron, A.; Liao, X.; Iglehart, J.D.; Livingston, D.M.; Ganesan, S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006, 9, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Dimitrov, S.D.; Feunteun, J.; Gelman, R.; Drapkin, R.; Lu, S.D.; Shestakova, E.; Velmurugan, S.; Denunzio, N.; Dragomir, S.; et al. Further evidence for BRCA1 communication with the inactive X chromosome. Cell 2007, 128, 991–1002. [Google Scholar] [CrossRef] [Green Version]
- Sirchia, S.M.; Ramoscelli, L.; Grati, F.R.; Barbera, F.; Coradini, D.; Rossella, F.; Porta, G.; Lesma, E.; Ruggeri, A.; Radice, P.; et al. Loss of the inactive X chromosome and replication of the active X in BRCA1-defective and wild-type breast cancer cells. Cancer Res. 2005, 65, 2139–2146. [Google Scholar] [CrossRef] [Green Version]
- Sirchia, S.M.; Tabano, S.; Monti, L.; Recalcati, M.P.; Gariboldi, M.; Grati, F.R.; Porta, G.; Finelli, P.; Radice, P.; Miozzo, M. Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE 2009, 4, e5559. [Google Scholar] [CrossRef]
- Vincent-Salomon, A.; Ganem-Elbaz, C.; Manie, E.; Raynal, V.; Sastre-Garau, X.; Stoppa-Lyonnet, D.; Stern, M.H.; Heard, E. X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 2007, 67, 5134–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, M.H.; Hudson, T.J.; Maire, G.; Squire, J.A.; Arcand, S.L.; Provencher, D.; Mes-Masson, A.M.; Tonin, P.N. Global analysis of chromosome X gene expression in primary cultuRes. of normal ovarian surface epithelial cells and epithelial ovarian cancer cell lines. Int. J. Oncol. 2007, 30, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, T.; Zhang, C.; Taniguchi, T.; Kim, C.J.; Okada, Y.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okamoto, K. Characterization of loss-of-inactive X in Klinefelter syndrome and female-derived cancer cells. Oncogene 2004, 23, 6163–6169. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx RNA activates Xist by evicting CTCF. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, T.S.; Loos, F.; van Staveren, S.; Myronova, E.; Ghazvini, M.; Grootegoed, J.A.; Gribnau, J. The trans-activator RNF12 and cis-acting elements effectuate X chromosome inactivation independent of X-pairing. Mol. Cell 2014, 53, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlan, G.; Gutierrez-Hernandez, N.; Huret, C.; Galupa, R.; van Bemmel, J.G.; Romito, A.; Heard, E.; Morey, C.; Rougeulle, C. The Ftx Noncoding Locus Controls X Chromosome Inactivation Independently of Its RNA Products. Mol. Cell 2018, 70, 462–472.e468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef]
- Shi, Y.; Downes, M.; Xie, W.; Kao, H.Y.; Ordentlich, P.; Tsai, C.C.; Hon, M.; Evans, R.M. Sharp, an inducible cofactor that integrates nuclear receptor repression and activation. Genes Dev. 2001, 15, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Moindrot, B.; Cerase, A.; Coker, H.; Masui, O.; Grijzenhout, A.; Pintacuda, G.; Schermelleh, L.; Nesterova, T.B.; Brockdorff, N. A Pooled shRNA Screen Identifies Rbm15, Spen, and Wtap as Factors Required for Xist RNA-Mediated Silencing. Cell Rep. 2015, 12, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Monfort, A.; di Minin, G.; Postlmayr, A.; Freimann, R.; Arieti, F.; Thore, S.; Wutz, A. Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep. 2015, 12, 554–561. [Google Scholar] [CrossRef] [Green Version]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Dossin, F.; Pinheiro, I.; Zylicz, J.J.; Roensch, J.; Collombet, S.; le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Apedaile, A.; Coker, H.; Termanis, A.; Zvetkova, I.; Godwin, J.; Tang, Y.A.; Huntley, D.; Montana, G.; Taylor, S.; et al. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 2012, 23, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.L.; Rigolet, M.; Bourc’his, D.; Nigon, F.; Bokesoy, I.; Fryns, J.P.; Hulten, M.; Jonveaux, P.; Maraschio, P.; Megarbane, A.; et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005, 25, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, E.; Lev, A.; Eyal, E.; Barel, O.; Kol, N.; Barhom, S.F.; Pode-Shakked, B.; Anikster, Y.; Somech, R.; Simon, A.J. A Novel Mutation in a Critical Region for the Methyl Donor Binding in DNMT3B Causes Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome (ICF). J. Clin. Immunol. 2016, 36, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Shirohzu, H.; Kubota, T.; Kumazawa, A.; Sado, T.; Chijiwa, T.; Inagaki, K.; Suetake, I.; Tajima, S.; Wakui, K.; Miki, Y.; et al. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002, 112, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjostrom, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Herold, T.; Metzeler, K.H.; Vosberg, S.; Hartmann, L.; Jurinovic, V.; Opatz, S.; Konstandin, N.P.; Schneider, S.; Zellmeier, E.; Ksienzyk, B.; et al. Acute myeloid leukemia with del(9q) is characterized by frequent mutations of NPM1, DNMT3A, WT1 and low expression of TLE4. Genes Chromosomes Cancer 2017, 56, 75–86. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Pintacuda, G.; Wei, G.; Roustan, C.; Kirmizitas, B.A.; Solcan, N.; Cerase, A.; Castello, A.; Mohammed, S.; Moindrot, B.; Nesterova, T.B.; et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 2017, 68, 955–969.e910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, M.Y.; Lammer, N.C.; Batey, R.T.; Wuttke, D.S. hnRNPK recognition of the B motif of Xist and other biological RNAs. Nucleic Acids Res. 2020, 48, 9320–9335. [Google Scholar] [CrossRef]
- Gallardo, M.; Lee, H.J.; Zhang, X.; Bueso-Ramos, C.; Pageon, L.R.; McArthur, M.; Multani, A.; Nazha, A.; Manshouri, T.; Parker-Thornburg, J.; et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell 2015, 28, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Au, P.Y.B.; Goedhart, C.; Ferguson, M.; Breckpot, J.; Devriendt, K.; Wierenga, K.; Fanning, E.; Grange, D.K.; Graham, G.E.; Galarreta, C.; et al. Phenotypic spectrum of Au-Kline syndrome: A report of six new cases and review of the literature. Eur. J. Hum. Genet. 2018, 26, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Torres, A.N.; Cats, D.; Mei, H.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Genomic analysis reveals recurrent deletion of JAK-STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer 2018, 57, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Dentici, M.L.; Barresi, S.; Niceta, M.; Pantaleoni, F.; Pizzi, S.; Dallapiccola, B.; Tartaglia, M.; Digilio, M.C. Clinical spectrum of Kabuki-like syndrome caused by HNRNPK haploinsufficiency. Clin. Genet. 2018, 93, 401–407. [Google Scholar] [CrossRef]
- Miyake, N.; Inaba, M.; Mizuno, S.; Shiina, M.; Imagawa, E.; Miyatake, S.; Nakashima, M.; Mizuguchi, T.; Takata, A.; Ogata, K.; et al. A case of atypical Kabuki syndrome arising from a novel missense variant in HNRNPK. Clin. Genet. 2017, 92, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Naarmann-de-Vries, I.S.; Sackmann, Y.; Klein, F.; Ostareck-Lederer, A.; Ostareck, D.H.; Jost, E.; Ehninger, G.; Brummendorf, T.H.; Marx, G.; Rollig, C.; et al. Characterization of acute myeloid leukemia with del(9q)—Impact of the genes in the minimally deleted region. Leuk Res. 2019, 76, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Maystadt, I.; Deprez, M.; Moortgat, S.; Benoit, V.; Karadurmus, D. A second case of Okamoto syndrome caused by HNRNPK mutation. Am. J. Med. Genet. A 2020, 182, 1537–1539. [Google Scholar] [CrossRef]
- Okamoto, N. Okamoto syndrome has featuRes. overlapping with Au-Kline syndrome and is caused by HNRNPK mutation. Am. J. Med. Genet. A 2019, 179, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, V.; Mocavini, I.; di Croce, L. Polycomb complexes in normal and malignant hematopoiesis. J. Cell Biol. 2019, 218, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef]
- Palau, A.; Garz, A.K.; Diesch, J.; Zwick, A.; Malinverni, R.; Valero, V.; Lappin, K.; Casquero, R.; Lennartsson, A.; Zuber, J.; et al. Polycomb protein RING1A limits hematopoietic differentiation in myelodysplastic syndromes. Oncotarget 2017, 8, 115002–115017. [Google Scholar] [CrossRef]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef]
- Underhill, C.; Qutob, M.S.; Yee, S.P.; Torchia, J. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 2000, 275, 40463–40470. [Google Scholar] [CrossRef] [Green Version]
- Pareja, F.; Ferrando, L.; Lee, S.S.K.; Beca, F.; Selenica, P.; Brown, D.N.; Farmanbar, A.; da Cruz-Paula, A.; Vahdatinia, M.; Zhang, H.; et al. The genomic landscape of metastatic histologic special types of invasive breast cancer. NPJ Breast Cancer 2020, 6, 53. [Google Scholar] [CrossRef]
- Nishi, A.; Numata, S.; Tajima, A.; Zhu, X.; Ito, K.; Saito, A.; Kato, Y.; Kinoshita, M.; Shimodera, S.; Ono, S.; et al. De novo non-synonymous TBL1XR1 mutation alters Wnt signaling activity. Sci. Rep. 2017, 7, 2887. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Yoo, H.Y.; Lee, S.H.; Shin, S.; Kim, S.C.; Lee, S.; Joung, J.G.; Nam, J.Y.; Ryu, D.; Yun, J.W.; et al. The mutational landscape of ocular marginal zone lymphoma identifies frequent alterations in TNFAIP3 followed by mutations in TBL1XR1 and CREBBP. Oncotarget 2017, 8, 17038–17049. [Google Scholar] [CrossRef] [Green Version]
- Heinen, C.A.; Jongejan, A.; Watson, P.J.; Redeker, B.; Boelen, A.; Boudzovitch-Surovtseva, O.; Forzano, F.; Hordijk, R.; Kelley, R.; Olney, A.H.; et al. A specific mutation in TBL1XR1 causes Pierpont syndrome. J. Med. Genet. 2016, 53, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz-Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Pons, L.; Cordier, M.P.; Labalme, A.; Till, M.; Louvrier, C.; Schluth-Bolard, C.; Lesca, G.; Edery, P.; Sanlaville, D. A new syndrome of intellectual disability with dysmorphism due to TBL1XR1 deletion. Am. J. Med. Genet. A 2015, 167A, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Tohyama, J.; Walsh, T.; Kato, M.; Kobayashi, Y.; Lee, M.; Tsurusaki, Y.; Miyake, N.; Goto, Y.; Nishino, I.; et al. A girl with West syndrome and autistic featuRes. harboring a de novo TBL1XR1 mutation. J. Hum. Genet. 2014, 59, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Riehmer, V.; Erger, F.; Herkenrath, P.; Seland, S.; Jackels, M.; Wiater, A.; Heller, R.; Beck, B.B.; Netzer, C. A heritable microduplication encompassing TBL1XR1 causes a genomic sister-disorder for the 3q26.32 microdeletion syndrome. Am. J. Med. Genet. A 2017, 173, 2132–2138. [Google Scholar] [CrossRef]
- Ivanov, I.; Lo, K.C.; Hawthorn, L.; Cowell, J.K.; Ionov, Y. Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene 2007, 26, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Sinha, R.; Hoadley, K.A.; Jacobsen, A.S.; Reva, B.; Perou, C.M.; Sander, C.; Schultz, N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res. Treat 2013, 141, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.R.; Huang, J.; Xu, Z.G.; Qian, B.Z.; Zhu, Z.D.; Yan, Q.; Cai, T.; Zhang, X.; Xiao, H.S.; Qu, J.; et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc. Natl. Acad. Sci. USA 2001, 98, 15089–15094. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Al-Dosari, M.S.; Al-Yacoub, N.; Colak, D.; Salih, M.A.; Alkuraya, F.S.; Poizat, C. Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum. Mol. Genet. 2013, 22, 2200–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.M.; Akunowicz, J.D.; Reveles, X.T.; Patel, B.B.; Saria, E.A.; Gorlick, R.G.; Naylor, S.L.; Leach, R.J.; Hansen, M.F. PHC3, a component of the hPRC-H complex, associates with E2F6 during G0 and is lost in osteosarcoma tumors. Oncogene 2007, 26, 1714–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Y.; Sun, S.X.; Zhu, S.X.; Bu, J. Identification of the Roles of Chromobox Family Members in Gastric Cancer: A Study Based on Multiple Datasets. Biomed. Res. Int. 2020, 2020, 5306509. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Lee, J.H.; Zhao, X.M.; Yoon, I.; Lee, J.Y.; Kwon, N.H.; Wang, Y.Y.; Lee, K.M.; Lee, M.J.; Kim, J.; Moon, H.G.; et al. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov. 2016, 2, 16025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, I.; Batty, E.M.; Pole, J.C.; Blood, K.A.; Mo, S.; Cooke, S.L.; Ng, C.; Howe, K.L.; Chin, S.F.; Brenton, J.D.; et al. Structural analysis of the genome of breast cancer cell line ZR-75-30 identifies twelve expressed fusion genes. BMC Genom. 2012, 13, 719. [Google Scholar] [CrossRef] [Green Version]
- Turnpenny, P.D.; Wright, M.J.; Sloman, M.; Caswell, R.; van Essen, A.J.; Gerkes, E.; Pfundt, R.; White, S.M.; Shaul-Lotan, N.; Carpenter, L.; et al. Missense Mutations of the Pro65 Residue of PCGF2 Cause a Recognizable Syndrome Associated with Craniofacial, Neurological, Cardiovascular, and Skeletal Features. Am. J. Hum. Genet. 2018, 103, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Kong, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatuRes. and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biason-Lauber, A.; Konrad, D.; Meyer, M.; DeBeaufort, C.; Schoenle, E.J. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. Am. J. Hum. Genet. 2009, 84, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, B.I.; Garcia, J.F.; Suela, J.; Mollejo, M.; Camacho, F.I.; Carro, A.; Montes, S.; Piris, M.A.; Cigudosa, J.C. Comparative genome profiling across subtypes of low-grade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica 2008, 93, 670–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Bodor, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Score, J.; Hidalgo-Curtis, C.; Jones, A.V.; Winkelmann, N.; Skinner, A.; Ward, D.; Zoi, K.; Ernst, T.; Stegelmann, F.; Dohner, K.; et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012, 119, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, S.; Takenobu, H.; Kageyama, H.; Koseki, H.; Ishii, T.; Nakazawa, A.; Tatezaki, S.; Nakagawara, A.; Kamijo, T. Polycomb group molecule PHC3 regulates polycomb complex composition and prognosis of osteosarcoma. Cancer Sci. 2010, 101, 1646–1652. [Google Scholar] [CrossRef]
- Brecqueville, M.; Cervera, N.; Adelaide, J.; Rey, J.; Carbuccia, N.; Chaffanet, M.; Mozziconacci, M.J.; Vey, N.; Birnbaum, D.; Gelsi-Boyer, V.; et al. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J. 2011, 1, e33. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.B.; Sun, S.L.; Zheng, Q.L.; Zhang, L.; Zhu, Y.; Jin, G.H.; Xue, L.X. Genetic alteration and misexpression of Polycomb group genes in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 2969–2979. [Google Scholar]
- Carter, A.C.; Xu, J.; Nakamoto, M.Y.; Wei, Y.; Zarnegar, B.J.; Shi, Q.; Broughton, J.P.; Ransom, R.C.; Salhotra, A.; Nagaraja, S.D.; et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. Elife 2020, 9. [Google Scholar] [CrossRef]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, J.B.; Lee, D.M.; Cherney, R.E.; Kim, S.O.; Inoue, K.; Schertzer, M.D.; Bischoff, S.R.; Cowley, D.O.; Calabrese, J.M. Elements at the 5’ end of Xist harbor SPEN-independent transcriptional antiterminator activity. Nucleic Acids Res. 2020, 48, 10500–10517. [Google Scholar] [CrossRef]
- Stephens, P.J.; Davies, H.R.; Mitani, Y.; van Loo, P.; Shlien, A.; Tarpey, P.S.; Papaemmanuil, E.; Cheverton, A.; Bignell, G.R.; Butler, A.P.; et al. Whole exome sequencing of adenoid cystic carcinoma. J. Clin. Investig. 2013, 123, 2965–2968. [Google Scholar] [CrossRef]
- Hansen, M.H.; Cedile, O.; Blum, M.K.; Hansen, S.V.; Ebbesen, L.H.; Bentzen, H.H.N.; Thomassen, M.; Kruse, T.A.; Kavan, S.; Kjeldsen, E.; et al. Molecular characterization of sorted malignant B cells from patients clinically identified with mantle cell lymphoma. Exp. Hematol. 2020, 84, 7–18.e12. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Zhang, S.; Kanagal-Shamanna, R.; Ok, C.Y.; Nomie, K.; Gonzalez, G.N.; Gonzalez-Pagan, O.; Hill, H.A.; Lee, H.J.; Fayad, L.; et al. Genomic profiles and clinical outcomes of de novo blastoid/pleomorphic MCL are distinct from those of transformed MCL. Blood Adv. 2020, 4, 1038–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic mutations and featuRes. of mantle cell lymphoma: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [Google Scholar] [CrossRef]
- Hartert, K.T.; Wenzl, K.; Krull, J.E.; Manske, M.; Sarangi, V.; Asmann, Y.; Larson, M.C.; Maurer, M.J.; Slager, S.; Macon, W.R.; et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia 2020. [Google Scholar] [CrossRef]
- Parry, M.; Rose-Zerilli, M.J.; Gibson, J.; Ennis, S.; Walewska, R.; Forster, J.; Parker, H.; Davis, Z.; Gardiner, A.; Collins, A.; et al. Whole exome sequencing identifies novel recurrently mutated genes in patients with splenic marginal zone lymphoma. PLoS ONE 2013, 8, e83244. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Fama, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Song, B.; Guo, S.; Li, G.; Jin, G. Identification of germline and somatic mutations in pancreatic adenosquamous carcinoma using whole exome sequencing. Cancer Biomark 2020, 27, 389–397. [Google Scholar] [CrossRef]
- Wang, T.; Hoekzema, K.; Vecchio, D.; Wu, H.; Sulovari, A.; Coe, B.P.; Gillentine, M.A.; Wilfert, A.B.; Perez-Jurado, L.A.; Kvarnung, M.; et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 2020, 11, 4932. [Google Scholar] [CrossRef]
- Zylicz, J.J.; Bousard, A.; Zumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197.e123. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Zhang, Y.; Xu, R.M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Brown, J.L.; Cao, R.; Zhang, Y.; Kassis, J.A.; Jones, R.S. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 2004, 14, 637–646. [Google Scholar] [CrossRef]
- Chaligne, R.; Heard, E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014, 588, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Pageau, G.J.; Hall, L.L.; Ganesan, S.; Livingston, D.M.; Lawrence, J.B. The disappearing Barr body in breast and ovarian cancers. Nat. Rev. Cancer 2007, 7, 628–633. [Google Scholar] [CrossRef]
- Brown, C.J.; Willard, H.F. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 1994, 368, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Csankovszki, G.; Panning, B.; Bates, B.; Pehrson, J.R.; Jaenisch, R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 1999, 22, 323–324. [Google Scholar] [CrossRef]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Kain, M.; Wang, L. Inactivation of X-linked tumor suppressor genes in human cancer. Future Oncol. 2012, 8, 463–481. [Google Scholar] [CrossRef] [Green Version]
- Spatz, A.; Borg, C.; Feunteun, J. X-chromosome genetics and human cancer. Nat. Rev. Cancer 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Chaligne, R.; Popova, T.; Mendoza-Parra, M.A.; Saleem, M.A.; Gentien, D.; Ban, K.; Piolot, T.; Leroy, O.; Mariani, O.; Gronemeyer, H.; et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res. 2015, 25, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Jager, N.; Schlesner, M.; Jones, D.T.; Raffel, S.; Mallm, J.P.; Junge, K.M.; Weichenhan, D.; Bauer, T.; Ishaque, N.; Kool, M.; et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell 2013, 155, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Dou, J.; Yang, G.; Chen, F. Long non-coding RNA XIST expression as a prognostic factor in human cancers: A meta-analysis. Int. J. Biol. Markers 2019, 34, 327–333. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Feng, L.; Li, F.; Sun, Z.; Wu, T.; Shi, X.; Li, J.; Li, X. Comprehensive characterization of lncRNA-mRNA related ceRNA network across 12 major cancers. Oncotarget 2016, 7, 64148–64167. [Google Scholar] [CrossRef] [Green Version]
- Madhi, H.; Kim, M.H. Beyond X-Chromosome Inactivation: The Oncogenic Facet of XIST in Human Cancers. Biomed. Sci. Lett. 2019, 25, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Giaimo, B.D.; Oswald, F.; Borggrefe, T. Dynamic chromatin regulation at Notch target genes. Transcription 2017, 8, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.T.; Su, H.; Khodadadi-Jamayran, A.; Lin, S.; Zhang, L.; Zhou, D.; Pawlik, K.M.; Townes, T.M.; Chen, Y.; Mulloy, J.C.; et al. The AS-RBM15 lncRNA enhances RBM15 protein translation during megakaryocyte differentiation. EMBO Rep. 2016, 17, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Zhang, J.; Breslin, P.; Onciu, M.; Ma, Z.; Morris, S.W. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 2009, 114, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Hiriart, E.; Gruffat, H.; Buisson, M.; Mikaelian, I.; Keppler, S.; Meresse, P.; Mercher, T.; Bernard, O.A.; Sergeant, A.; Manet, E. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1, and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J. Biol. Chem. 2005, 280, 36935–36945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, H.; Wei, G.; Moindrot, B.; Mohammed, S.; Nesterova, T.; Brockdorff, N. The role of the Xist 5’ m6A region and RBM15 in X chromosome inactivation. Wellcome Open Res. 2020, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Newberry, E.P.; Latifi, T.; Towler, D.A. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry 1999, 38, 10678–10690. [Google Scholar] [CrossRef]
- Oswald, F.; Kostezka, U.; Astrahantseff, K.; Bourteele, S.; Dillinger, K.; Zechner, U.; Ludwig, L.; Wilda, M.; Hameister, H.; Knochel, W.; et al. SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J. 2002, 21, 5417–5426. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Vander-Wielen, B.D.; Giaimo, B.D.; Pan, L.; Collins, C.E.; Turkiewicz, A.; Hein, K.; Oswald, F.; Borggrefe, T.; Kovall, R.A. Structural and Functional Studies of the RBPJ-SHARP Complex Reveal a Conserved Corepressor Binding Site. Cell Rep. 2019, 26, 845–854.e846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, F.; Rodriguez, P.; Giaimo, B.D.; Antonello, Z.A.; Mira, L.; Mittler, G.; Thiel, V.N.; Collins, K.J.; Tabaja, N.; Cizelsky, W.; et al. A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucleic Acids Res. 2016, 44, 4703–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, F.; Winkler, M.; Cao, Y.; Astrahantseff, K.; Bourteele, S.; Knochel, W.; Borggrefe, T. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol. Cell Biol. 2005, 25, 10379–10390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salat, D.; Liefke, R.; Wiedenmann, J.; Borggrefe, T.; Oswald, F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-Jkappa/SHARP-mediated repression of notch target genes. Mol. Cell Biol. 2008, 28, 3502–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, V.N.; Giaimo, B.D.; Schwarz, P.; Soller, K.; Vas, V.; Bartkuhn, M.; Blatte, T.J.; Dohner, K.; Bullinger, L.; Borggrefe, T.; et al. Heterodimerization of AML1/ETO with CBFbeta is required for leukemogenesis but not for myeloproliferation. Leukemia 2017, 31, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Han, H.; Tani, S.; Tanigaki, K.; Tun, T.; Furukawa, T.; Taniguchi, Y.; Kurooka, H.; Hamada, Y.; Toyokuni, S.; et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity 2003, 18, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.; Shinkura, R.; Kuroda, K.; Yabe, D.; Honjo, T. Msx2-interacting nuclear target protein (Mint) deficiency reveals negative regulation of early thymocyte differentiation by Notch/RBP-J signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Sierra, O.L.; Cheng, S.L.; Loewy, A.P.; Charlton-Kachigian, N.; Towler, D.A. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J. Biol. Chem. 2004, 279, 32913–32923. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, J.; Qin, H.; Yang, H.; Li, J.; Zhou, P.; Liang, Y.; Han, H. Mint represses transactivation of the type II collagen gene enhancer through interaction with alpha A-crystallin-binding protein 1. J. Biol. Chem. 2005, 280, 18710–18716. [Google Scholar] [CrossRef] [Green Version]
- Giaimo, B.D.; Borggrefe, T. Introduction to Molecular Mechanisms in Notch Signal Transduction and Disease Pathogenesis. Adv. Exp. Med. Biol. 2018, 1066, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, B.D.; Gagliani, E.K.; Kovall, R.A.; Borggrefe, T. Transcription Factor RBPJ as a Molecular Switch in Regulating the Notch Response. Adv. Exp. Med. Biol. 2021, 1287, 9–30. [Google Scholar] [CrossRef]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar] [CrossRef]
- Arieti, F.; Gabus, C.; Tambalo, M.; Huet, T.; Round, A.; Thore, S. The crystal structure of the Split End protein SHARP adds a new layer of complexity to proteins containing RNA recognition motifs. Nucleic Acids Res. 2014, 42, 6742–6752. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger, LLC. The PyMOL. Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFbeta/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Hayward, S.D. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell Biol. 2001, 21, 6222–6232. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Fujimuro, M.; Hsieh, J.J.; Chen, L.; Miyamoto, A.; Weinmaster, G.; Hayward, S.D. SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC To facilitate NotchIC function. Mol. Cell Biol. 2000, 20, 2400–2410. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Fujimuro, M.; Hsieh, J.J.; Chen, L.; Hayward, S.D. A role for SKIP in EBNA2 activation of CBF1-repressed promoters. J. Virol. 2000, 74, 1939–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Choi, H.K.; Choi, K.C.; Park, S.Y.; Ota, I.; Yook, J.I.; Lee, Y.H.; Kim, K.; Yoon, H.G. Nuclear hormone receptor corepressor promotes esophageal cancer cell invasion by transcriptional repression of interferon-gamma-inducible protein 10 in a casein kinase 2-dependent manner. Mol. Biol. Cell 2012, 23, 2943–2954. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Lim, B.J.; Choi, H.K.; Hong, S.W.; Jang, H.S.; Kim, C.; Chun, K.H.; Choi, K.C.; Yoon, H.G. CK2-NCoR signaling cascade promotes prostate tumorigenesis. Oncotarget 2013, 4, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gross, W.; Hong, S.H.; Privalsky, M.L. The SMRT corepressor is a target of phosphorylation by protein kinase CK2 (casein kinase II). Mol. Cell Biochem. 2001, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vander-Wielen, B.D.; Yuan, Z.; Friedmann, D.R.; Kovall, R.A. Transcriptional repression in the Notch pathway: Thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J. Biol. Chem. 2011, 286, 14892–14902. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, M.; Schwabe, J.W. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev. 2003, 17, 1909–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villasenor, R.; Hess, D.; et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, J.; Yang, X.; Li, J.; Qin, H.; Dong, X.; Zhu, Y.; Liang, L.; Liang, Y.; Han, H. The Spen homolog Msx2-interacting nuclear target protein interacts with the E2 ubiquitin-conjugating enzyme UbcH8. Mol. Cell Biochem. 2006, 288, 151–157. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Yang, X.; Qin, H.; Zhou, P.; Liang, Y.; Han, H. The C terminus of MINT forms homodimers and abrogates MINT-mediated transcriptional repression. Biochim. Biophys. Acta 2005, 1729, 50–56. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.G.; Chan, D.W.; Huang, Z.Q.; Li, J.; Fondell, J.D.; Qin, J.; Wong, J. Purification and functional characterization of the human N-CoR complex: The roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003, 22, 1336–1346. [Google Scholar] [CrossRef]
- Yoon, H.G.; Chan, D.W.; Reynolds, A.B.; Qin, J.; Wong, J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 2003, 12, 723–734. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, M.T.; Ramesar, R.S.; Caciotti, B.; Winship, I.M.; de Grandi, A.; Riboni, M.; Townes, P.L.; Beighton, P.; Ballabio, A.; Borsani, G. X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. Am. J. Hum. Genet. 1999, 64, 1604–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codina, A.; Love, J.D.; Li, Y.; Lazar, M.A.; Neuhaus, D.; Schwabe, J.W. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2005, 102, 6009–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, F.; Giaimo, B.D.; Bartkuhn, M.; Zimmermann, T.; Close, V.; Mertens, D.; Nist, A.; Stiewe, T.; Meier-Soelch, J.; Kracht, M.; et al. HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res. 2020, 48, 3496–3512. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [Green Version]
- You, S.H.; Lim, H.W.; Sun, Z.; Broache, M.; Won, K.J.; Lazar, M.A. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 2013, 20, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Barish, G.D.; Yu, R.T.; Karunasiri, M.S.; Becerra, D.; Kim, J.; Tseng, T.W.; Tai, L.J.; Leblanc, M.; Diehl, C.; Cerchietti, L.; et al. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012, 15, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.F.; Melnick, A.; Lax, S.; Bouchard, D.; Liu, J.; Kiang, C.L.; Mayer, S.; Takahashi, S.; Licht, J.D.; Prive, G.G. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 2003, 12, 1551–1564. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Gaudet, J.; Cheney, M.D.; Roudaia, L.; Cierpicki, T.; Klet, R.C.; Hartman, K.; Laue, T.M.; Speck, N.A.; et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO’s activity. Cancer Cell 2007, 11, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Huang, W.; Jepsen, K.; Benner, C.; Hardiman, G.; Rosenfeld, M.G.; Glass, C.K. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009, 23, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Lazar, M.A. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 2005, 19, 1452–1459. [Google Scholar] [CrossRef]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, S.; Kanaba, T.; Ito, Y.; Mishima, M. NMR assignments of SPOC domain of the human transcriptional corepressor SHARP in complex with a C-terminal SMRT peptide. BioMol. NMR Assign. 2013, 7, 267–270. [Google Scholar] [CrossRef]
- Mikami, S.; Kanaba, T.; Takizawa, N.; Kobayashi, A.; Maesaki, R.; Fujiwara, T.; Ito, Y.; Mishima, M. Structural insights into the recruitment of SMRT by the corepressor SHARP under phosphorylative regulation. Structure 2014, 22, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Vadlamudi, R.K.; Manavathi, B.; Singh, R.R.; Nguyen, D.; Li, F.; Kumar, R. An essential role of Pak1 phosphorylation of SHARP in Notch signaling. Oncogene 2005, 24, 4591–4596. [Google Scholar] [CrossRef] [Green Version]
- Legare, S.; Cavallone, L.; Mamo, A.; Chabot, C.; Sirois, I.; Magliocco, A.; Klimowicz, A.; Tonin, P.N.; Buchanan, M.; Keilty, D.; et al. The Estrogen Receptor Cofactor SPEN Functions as a Tumor Suppressor and Candidate Biomarker of Drug Responsiveness in Hormone-Dependent Breast Cancers. Cancer Res. 2015, 75, 4351–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Bommer, G.T.; Zhai, Y.; Akyol, A.; Hinoi, T.; Winer, I.; Lin, H.V.; Cadigan, K.M.; Cho, K.R.; Fearon, E.R. Drosophila split ends homologue SHARP functions as a positive regulator of Wnt/beta-catenin/T-cell factor signaling in neoplastic transformation. Cancer Res. 2007, 67, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansithong, W.; Jog, S.P.; Paul, S.; Mohammadzadeh, R.; Tring, S.; Kwok, Y.; Fry, R.C.; Marjoram, P.; Comai, L.; Reddy, S. RNA steady-state defects in myotonic dystrophy are linked to nuclear exclusion of SHARP. EMBO Rep. 2011, 12, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Koshimizu, E.; Okamoto, N.; Mizuno, S.; Ogata, T.; Nagai, T.; Kosho, T.; Ohashi, H.; Kato, M.; Sasaki, G.; et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. A 2013, 161, 2234–2243. [Google Scholar] [CrossRef]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef]
- Micale, L.; Augello, B.; Fusco, C.; Selicorni, A.; Loviglio, M.N.; Silengo, M.C.; Reymond, A.; Gumiero, B.; Zucchetti, F.; D’Addetta, E.V.; et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulussen, A.D.; Stegmann, A.P.; Blok, M.J.; Tserpelis, D.; Posma-Velter, C.; Detisch, Y.; Smeets, E.E.; Wagemans, A.; Schrander, J.J.; van den Boogaard, M.J.; et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011, 32, E2018–E2025. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, M.C.; Buckingham, K.J.; Ng, S.B.; Ming, J.E.; Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Bigham, A.W.; Tabor, H.K.; Mefford, H.C.; et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A 2011, 155A, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bogershausen, N.; Alanay, Y.; Simsek-Kiper, P.O.; Plume, N.; Keupp, K.; Pohl, E.; Pawlik, B.; Rachwalski, M.; Milz, E.; et al. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011, 130, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; van Bon, B.W.; Steehouwer, M.; Rodriguez-Santiago, B.; Simpson, M.; Dias, P.; Anderlid, B.M.; Arts, P.; Bhat, M.; Augello, B.; et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: A genotype-phenotype study. Clin. Genet. 2013, 84, 539–545. [Google Scholar] [CrossRef]
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013, 34, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.L.A.; Demarest, B.L.; Tone-Pah-Hote, T.; Tristani-Firouzi, M.; Yost, H.J. Inhibition of Notch signaling rescues cardiovascular development in Kabuki Syndrome. PLoS Biol. 2019, 17, e3000087. [Google Scholar] [CrossRef] [PubMed]
- Mercher, T.; Coniat, M.B.; Monni, R.; Mauchauffe, M.; Nguyen-Khac, F.; Gressin, L.; Mugneret, F.; Leblanc, T.; Dastugue, N.; Berger, R.; et al. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 5776–5779. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Morris, S.W.; Valentine, V.; Li, M.; Herbrick, J.A.; Cui, X.; Bouman, D.; Li, Y.; Mehta, P.K.; Nizetic, D.; et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 2001, 28, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, S.; Zhang, Y.; Zhu, X. Biological effects of decreasing RBM15 on chronic myelogenous leukemia cells. Leuk Lymphoma 2012, 53, 2237–2244. [Google Scholar] [CrossRef]
- Kennison, J.A. Introduction to Trx-G and Pc-G genes. Methods EnzyMol. 2004, 377, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Wang, Y.; Jacobs, S.A.; Kim, Y.; Allis, C.D.; Khorasanizadeh, S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17, 1870–1881. [Google Scholar] [CrossRef] [Green Version]
- Schertzer, M.D.; Braceros, K.C.A.; Starmer, J.; Cherney, R.E.; Lee, D.M.; Salazar, G.; Justice, M.; Bischoff, S.R.; Cowley, D.O.; Ariel, P.; et al. lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol. Cell 2019, 75, 523–537.e510. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark Insights 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Menafra, R.; Stunnenberg, H.G. MBD2 and MBD3: Elusive functions and mechanisms. Front. Genet. 2014, 5, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voo, K.S.; Carlone, D.L.; Jacobsen, B.M.; Flodin, A.; Skalnik, D.G. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shaRes. a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell Biol. 2000, 20, 2108–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein/Complex | Subunits | Function(s) in XCI | Disease(s) | References |
|---|---|---|---|---|
| DNMT3B | - | DNA methyltransferase | AML, FSHD, HD, ICF, PR | [56,57,58,59,60,61,62,63,64] |
| hnRNPK | - | Bridging protein between Xist and ncPRC1 | AKS, AML, KLS, KS, MF, OS | [65,66,67,68,69,70,71,72,73,74,75] |
| ncPRC1 | PCGF3/5 RING1A/B RYBP/YAF | E3 ubiquitin ligase | MDS | [65,76,77,78] |
| NCoR1/2 * | GPS2 HDAC3 NCoR1/2 * TBL1 TBLR1 | Deacetylase | ASDs, BC, CC, HCC, ID, MB, NDDs, OMZL, PS, SCZ | [79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95] |
| PRC1 | CBX2/4/6/7/8 PCGF1-6 PHC1-3 RING1A/B SCMH1/L2 | E3 ubiquitin ligase/Recognition of histone methylation | BC, DD, DSD, ESCC, GC, MCL, MDS, OSS, PM | [76,78,96,97,98,99,100,101,102,103,104,105] |
| PRC2 | AEBP2 EZH2 ** EED JARID2 RBBP4/7 SUZ12 | Methyltransferase | AML, DS-AMKL, DLBCL, ETP-ALL, FL, HCC, MDS, MPN, T-ALL, T-PLL | [76,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127] |
| SHARP | - | Adaptor protein that recruits the HDAC3-containing NCoR1/2 complexes | ACC, BC, DLBCL, MCL, NDDs, PASC, SMZL | [51,52,53,54,55,128,129,130,131,132,133,134,135,136,137,138,139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giaimo, B.D.; Robert-Finestra, T.; Oswald, F.; Gribnau, J.; Borggrefe, T. Chromatin Regulator SPEN/SHARP in X Inactivation and Disease. Cancers 2021, 13, 1665. https://doi.org/10.3390/cancers13071665
Giaimo BD, Robert-Finestra T, Oswald F, Gribnau J, Borggrefe T. Chromatin Regulator SPEN/SHARP in X Inactivation and Disease. Cancers. 2021; 13(7):1665. https://doi.org/10.3390/cancers13071665
Chicago/Turabian StyleGiaimo, Benedetto Daniele, Teresa Robert-Finestra, Franz Oswald, Joost Gribnau, and Tilman Borggrefe. 2021. "Chromatin Regulator SPEN/SHARP in X Inactivation and Disease" Cancers 13, no. 7: 1665. https://doi.org/10.3390/cancers13071665
APA StyleGiaimo, B. D., Robert-Finestra, T., Oswald, F., Gribnau, J., & Borggrefe, T. (2021). Chromatin Regulator SPEN/SHARP in X Inactivation and Disease. Cancers, 13(7), 1665. https://doi.org/10.3390/cancers13071665