
Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunomodulatory Drugs
2.1. IMiD Mechanism of Action
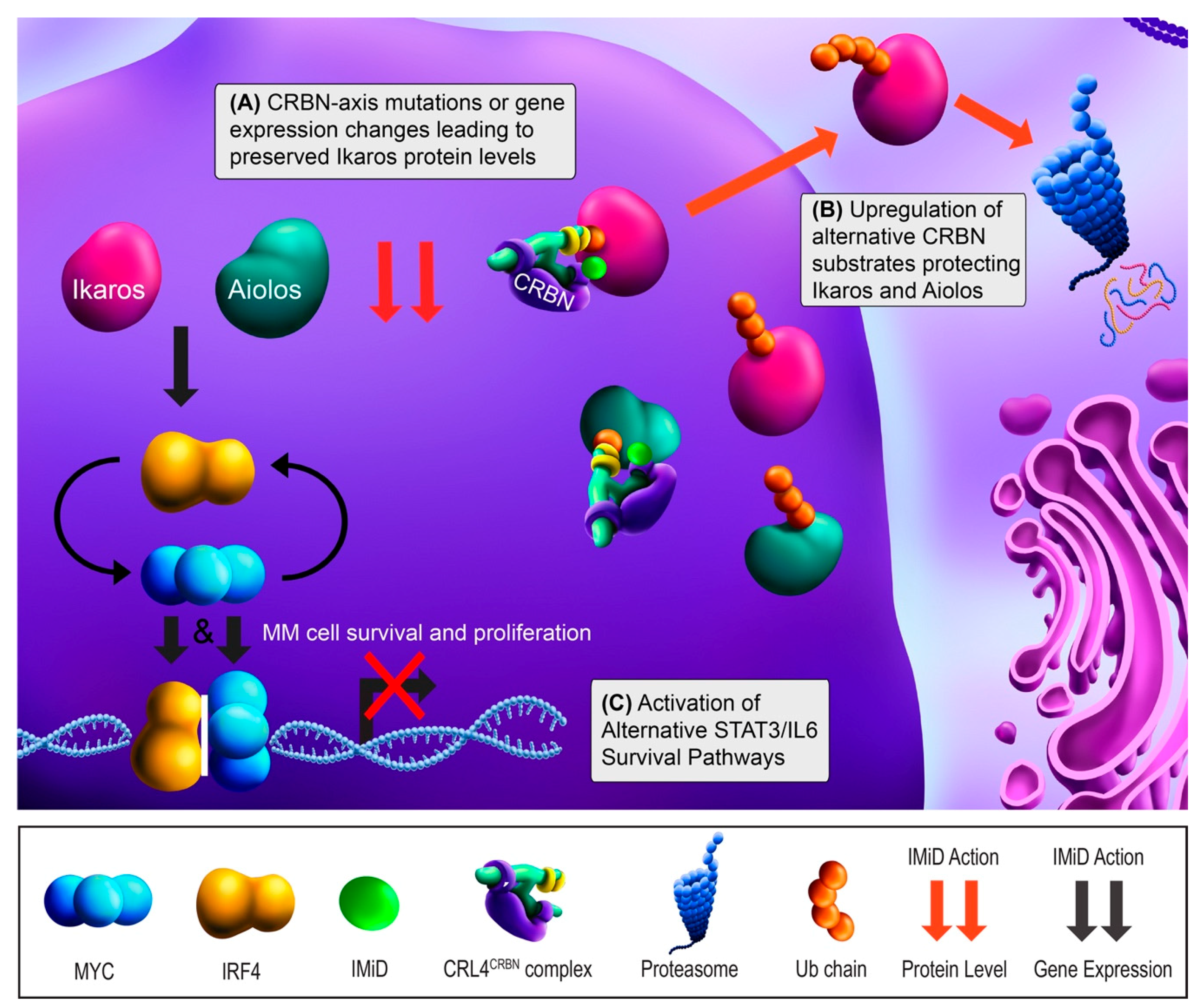
2.2. IMiD Resistance
2.3. Overcoming IMiD Resistance
3. Proteasome Inhibitors
3.1. PI Mechanism of Action
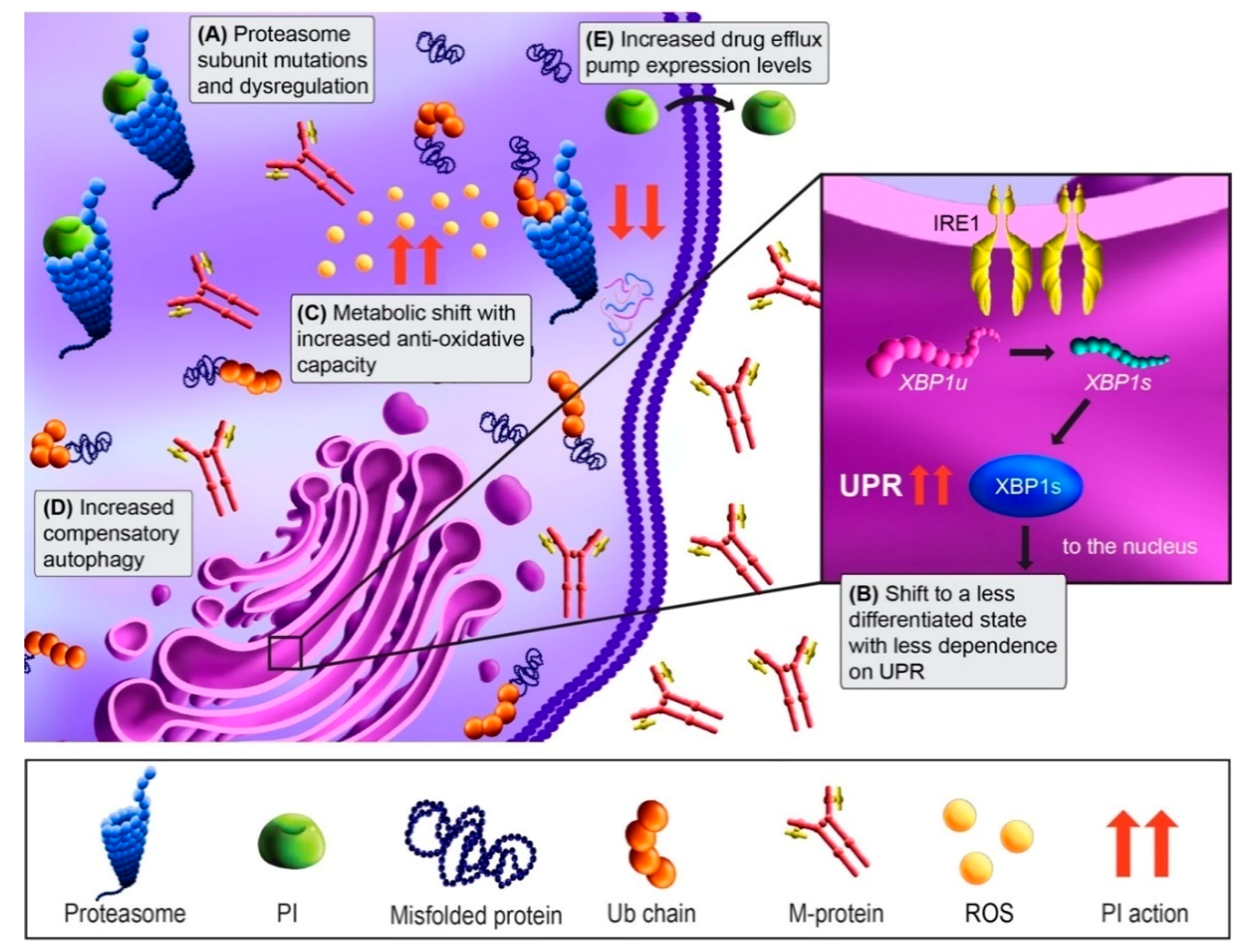
3.2. PI Resistance
3.3. Overcoming PI Resistance
4. Immunotherapies
4.1. Monoclonal Antibody Mechanism of Action
4.2. Immunotherapy Resistance
4.3. Overcoming Immunotherapy Resistance
5. Other Drug Classes
5.1. Dexamethasone Resistance
5.2. Melphalan Resistance
5.3. Overcoming Melphalan Resistance
6. Other Strategies for Circumventing Drug Resistance
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Van De Donk, N.W.C.J.; Zweegman, S.; Lokhorst, H.M. Current and New Therapeutic Strategies for Relapsed and Refractory Multiple Myeloma: An Update. Drugs 2018, 78, 19–37. [Google Scholar] [CrossRef] [Green Version]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Sperling, A.S.; Burgess, M.; Keshishian, H.; Gasser, J.A.; Bhatt, S.; Jan, M.; Słabicki, M.; Sellar, R.S.; Fink, E.C.; Miller, P.G.; et al. Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 2019, 134, 160–170. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; De Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Barrio, S.; Munawar, U.; Zhu, Y.X.; Giesen, N.; Shi, C.X.; Da Via, M.; Sanchez, R.; Bruins, L.; Demler, T.; Muller, N.; et al. IKZF1/3 and CRL4-CRBN E3 ubiquitin ligase mutations and IMiD resistance in multiple myeloma. Haematologica 2019, 105, e237–e241. [Google Scholar] [CrossRef] [Green Version]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, K.; Munch-Petersen, H.F.; Eskelund, C.W.; Sjö, L.D.; Ralfkiaer, E.; Gimsing, P.; Grønbaek, K. Expression of CRBN, IKZF1, and IKZF3 does not predict lenalidomide sensitivity and mutations in the cereblon pathway are infrequent in multiple myeloma. Leuk. Lymphoma 2018, 60, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Fernández-Lázaro, D.; San-Segundo, L.; López-Corral, L.; Sánchez, L.A.C.; Gutiérrez, N.C.; Garayoa, M.; Paíno, T.; Garcia-Gomez, A.; Delgado, M.; et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia 2014, 29, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Gutierrez-Uzquiza, A.; Zheng, X.Y.; Chang, R.; Vogl, D.T.; Garfall, A.L.; Bernabei, L.; Saraf, A.; Florens, L.; Washburn, M.P.; et al. RUNX proteins desensitize multiple myeloma to lenalidomide via protecting IKZFs from degradation. Leukemia 2019, 33, 2006–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Song, T.; Zhou, W.; Xing, L.; Wang, S.; Ho, M.; Peng, Z.; Tai, Y.-T.; Hideshima, T.; Anderson, K.C.; et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; Van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Shi, L.; Wang, S.; Zangari, M.; Xu, H.; Cao, T.M.; Xu, C.; Wu, Y.; Xiao, F.; Liu, Y.; Yang, Y.; et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010, 1, 22–33. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; Dulk, H.D.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Jing, X.; Holman, C.; Sompallae, R.; Zhan, F.; Tricot, G.; Yang, Y.; Janz, S. Upregulation of FOXM1 leads to diminished drug sensitivity in myeloma. BMC Cancer 2018, 18, 1152. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; Van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; Van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 helicase is involved in replication stress survival and drug resistance in multiple myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.; Hideshima, T.; Anderson, K. Thalidomide in multiple myeloma. Biomed. Pharmacother. 2002, 56, 115–128. [Google Scholar] [CrossRef]
- Sehgal, K.; Das, R.; Zhang, L.; Verma, R.; Deng, Y.; Kocoglu, M.; Vasquez, J.; Koduru, S.; Ren, Y.; Wang, M.; et al. Clinical and pharmacodynamic analysis of pomalidomide dosing strategies in myeloma: Impact of immune activation and cereblon targets. Blood 2015, 125, 4042–4051. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; Suman, V.J.; McCarthy, P.L. Update on the role of lenalidomide in patients with multiple myeloma. Ther. Adv. Hematol. 2018, 9, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Girona, A.; Mendy, D.; Ito, T.A.; Miller, K.H.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.E.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Ito, T.; Handa, H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int. J. Hematol. 2016, 104, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Słabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thomä, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Jr, W.G.K. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, L.B.; Ward, A.C. The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Mol. Immunol. 2011, 48, 1272–1278. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.S.; Breider, M.; et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nat. Cell Biol. 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Richardson, P.; Jagannath, S.; Hussein, M.; Berenson, J.; Singhal, S.; Irwin, D.; Williams, S.F.; Bensinger, W.; Badros, A.Z.; Vescio, R.; et al. Safety and efficacy of single-agent lenalidomide in patients with relapsed and refractory multiple myeloma. Blood 2009, 114, 772–778. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Gooding, S.; Ansari-Pour, N.; Towfic, F.; Estévez, M.O.; Chamberlain, P.P.; Tsai, K.-T.; Flynt, E.; Hirst, M.; Rozelle, D.; Dhiman, P.; et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood 2021, 137, 232–237. [Google Scholar] [CrossRef]
- Broyl, A.; Kuiper, R.; Van Duin, M.; Van Der Holt, B.; El Jarari, L.; Bertsch, U.; Zweegman, S.; Buijs, A.; Hose, D.; Lokhorst, H.M.; et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood 2013, 121, 624–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.R.; Kortuem, K.M.; Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Ahmann, G.; Kumar, S.; Rajkumar, S.V.; et al. The clinical significance of cereblon expression in multiple myeloma. Leuk. Res. 2014, 38, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Nijhof, I.S.; Couto, S.; Levin, M.-D.; Bos, G.M.; Broijl, A.; Klein, S.K.; Ren, Y.; Wang, M.; Koene, H.R.; et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematology 2018, 103, e368–e371. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Lin, C.-W.; Lin, H.-H.; Yao, M.; Tang, J.-L.; Wu, S.-J.; Chen, Y.-C.; Lu, H.-Y.; Hou, H.-A.; Chen, C.-Y.; et al. Expression of cereblon protein assessed by immunohistochemicalstaining in myeloma cells is associated with superior response of thalidomide- and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. Ann. Hematol. 2014, 93, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Kuchenbauer, F.; Kull, M.; Teleanu, V.; Bullinger, L.; Bunjes, D.; Greiner, A.; Kolmus, S.; Köpff, S.; Schreder, M.; et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: A study of the German Myeloma Study Group (DSMM). Leukemia 2016, 31, 1363–1367. [Google Scholar] [CrossRef]
- Yamamoto, J.; Suwa, T.; Murase, Y.; Tateno, S.; Mizutome, H.; Asatsuma-Okumura, T.; Shimizu, N.; Kishi, T.; Momose, S.; Kizaki, M.; et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat. Chem. Biol. 2020, 16, 1208–1217. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Zhang, W.; Man, H.-W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; Lebrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2017, 61, 535–542. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Van De Donk, N.W.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8006. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.F.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2018, 25, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, Z.J.; VanWyngarden, M.J.; Stevens, B.M.; Abbott, D.; Hammes, A.; Langouët-Astrie, C.; Smith, C.A.; Palmer, B.E.; Forsberg, P.A.; Mark, T.M.; et al. Measurement of ex vivo resistance to proteasome inhibitors, IMiDs, and daratumumab during multiple myeloma progression. Blood Adv. 2020, 4, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood 2016, 127, 2693–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Madduri, D.; Berdeja, J.G.; Usmani, M.S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, M.A.K.; Hari, M.P.; Htut, M.; O’Donnell, E.; et al. CARTITUDE-1: Phase 1b/2 Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 22–25. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; De La Rubia, J.; Mateos, M.-V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell–redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Cohen, A.D.; Trudel, S.; Forsberg, P.A.; Fonseca, R.; Krishnan, A.Y.; Spencer, A.; Berdeja, J.G.; Hamirani, S.M.; Li, M.; Cooper, J.; et al. GO39775: A multicenter phase I trial evaluating the safety, pharmacokinetics, and activity of BFCR4350A, a FcRH5/CD3 T-cell dependent bispecific antibody, in patients with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2020, 38, TPS8551. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- VanWyngarden, M.; Walker, Z.; Su, Y.; Bearrows, S.; Stevens, B.; Forsberg, P.; Mark, T.; Smith, C.; Matsui, W.; Jordan, C.; et al. CD46 Antibody Drug Conjugate Impedes Myeloma Engraftment in Patient-Derived Xenografts. Clin. Lymphoma Myeloma Leuk. 2019, 19, e151. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Delimpasi, S.; Simonova, M.; Spicka, I.; Pour, L.; Kryachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; et al. Weekly selinexor, bortezomib, and dexamethasone (SVd) versus twice weekly bortezomib and dexamethasone (Vd) in patients with multiple myeloma (MM) after one to three prior therapies: Initial results of the phase III BOSTON study. J. Clin. Oncol. 2020, 38, 8501. [Google Scholar] [CrossRef]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.-T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351.e3. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Sokol, E.; Jin, D.; Brune, Z.; Thiru, P.; Ghandi, M.; Garraway, L.A.; Gupta, P.B.; Santagata, S.; Whitesell, L.; et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. USA 2016, 114, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promote Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef]
- Harnoss, J.M.; Le Thomas, A.; Shemorry, A.; Marsters, S.A.; Lawrence, D.A.; Lu, M.; Chen, Y.-C.A.; Qing, J.; Totpal, K.; Kan, D.; et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 16420–16429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef]
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358–12370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riz, I.; Hawley, T.S.; Hawley, R.G. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015, 6, 14814–14831. [Google Scholar] [CrossRef] [Green Version]
- Besse, A.; Besse, A.; Stolze, S.C.; Stolze, S.C.; Rasche, L.; Rasche, L.; Weinhold, N.; Weinhold, N.; Morgan, G.J.; Morgan, G.J.; et al. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 Induces Drug Resistance Mainly through Activation of Efflux Drug Pumps and Is Associated with Poor Prognosis in Myeloma and Other Cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2014, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, W.; Xia, J.; Gu, Z.; Wendlandt, E.; Zhan, X.; Janz, S.; Tricot, G.; Zhan, F. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget 2014, 5, 11986–11997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.K.; Odzorig, T.; Jin, W.; Xia, D. Structural Basis of p97 Inhibition by the Site-Selective Anticancer Compound Compound CB-5083. Mol. Pharmacol. 2019, 95, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.-T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp. Ther. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, Z.J.; Idler, B.M.; Davis, L.N.; Stevens, B.M.; VanWyngarden, M.J.; Ohlstrom, D.; Bearrows, S.C.; Hammes, A.; Smith, C.A.; Jordan, C.T.; et al. Exploiting Protein Translation Dependence in Multiple Myeloma with Omacetaxine-Based Therapy. Clin. Cancer Res. 2021, 27, 819–830. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; San-Miguel, J.; Belch, A.; White, D.; Benboubker, L.; Cook, G.; Leiba, M.; Morton, J.; Ho, P.J.; Kim, K.; et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of POLLUX. Haematology 2018, 103, 2088–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- De Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2010, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; Van Egmond, M.; Van Bueren, J.J.L.; Mutis, T.; Groen, R.W.J.; Breij, E.; Martens, A.C.M.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcgamma Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Dimopoulos, M.A.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Acha, O.P.D.; Idler, B.M.; Walker, Z.J.; Forsberg, P.A.; Mark, T.M.; Sherbenou, D.W. Myeloma Drug Sensitivity Testing to Optimize Retreatment with Anti-CD38 Monoclonal Antibodies in Daratumumab-Refractory Patients. Blood 2020, 136, 36–37. [Google Scholar]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.C.; Anderson, J.L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Raje, N.S.; Jakubowiak, A.; Gasparetto, C.; Cornell, R.F.; Krupka, H.I.; Navarro, D.; Forgie, A.J.; Udata, C.; Basu, C.; Chou, J.; et al. Safety, Clinical Activity, Pharmacokinetics, and Pharmacodynamics from a Phase I Study of PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 1869. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kroenke, J.; Facon, T.; Salnikov, A.; Lesley, R.; et al. Evaluation of AMG 420, an anti-BCMA bispecific T-cell engager (BiTE) immunotherapy, in R/R multiple myeloma (MM) patients: Updated results of a first-in-human (FIH) phase I dose escalation study. J. Clin. Oncol. 2019, 37, 8007. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Investig. 2016, 126, 4640–4653. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell–redirecting bispecific G-protein–coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Kervoëlen, C.; Ménoret, E.; Gomez-Bougie, P.; Bataille, R.; Godon, C.; Marionneau-Lambot, S.; Moreau, P.; Pellat-Deceunynck, C.; Amiot, M. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. Oncotarget 2015, 6, 26922–26934. [Google Scholar] [CrossRef] [Green Version]
- Gkotzamanidou, M.; Terpos, E.; Bamia, C.; Munshi, N.C.; Dimopoulos, M.A.; Souliotis, V.L. DNA repair of myeloma plasma cells correlates with clinical outcome: The effect of the nonhomologous end-joining inhibitor SCR7. Blood 2016, 128, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickström, M.; Nygren, P.; Larsson, R.; Harmenberg, J.; Lindberg, J.; Sjöberg, P.; Jerling, M.; Lehmann, F.; Richardson, P.; Anderson, K.; et al. Melflufen—A peptidase-potentiated alkylating agent in clinical trials. Oncotarget 2017, 8, 66641–66655. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; Azab, F.; De La Puente, P.; Landesman, Y.; Azab, A.K. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl. Oncol. 2017, 10, 632–640. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Touzeau, C.; Ryan, J.; Guerriero, J.L.; Moreau, P.; Ni Chonghaile, T.; Le Gouill, S.; Richardson, P.G.; Anderson, K.C.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, B.; Han, H.; Song, S.; Xu, H.; Hong, Y.; Yi, N.; Zhuang, W. Predicting multi-level drug response with gene expression profile in multiple myeloma using hierarchical ordinal regression. BMC Cancer 2018, 18, 551. [Google Scholar] [CrossRef] [Green Version]
- Cetin, A.E.; Stevens, M.M.; Calistri, N.L.; Fulciniti, M.; Olcum, S.A.; Kimmerling, R.J.; Munshi, N.C.; Manalis, S.R. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Stessman, H.A.; Baughn, L.B.; Sarver, A.; Xia, T.; Deshpande, R.; Mansoor, A.; Walsh, S.A.; Sunderland, J.J.; Dolloff, N.G.; Linden, M.A.; et al. Profiling Bortezomib Resistance Identifies Secondary Therapies in a Mouse Myeloma Model. Mol. Cancer Ther. 2013, 12, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Kikukawa, Y.; Fujiwara, S.; Wada, N.; Okuno, Y.; Mitsuya, H.; Hata, H. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int. J. Oncol. 2013, 43, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Drug Name | Drug MOA | Resistance Mechanisms | Refs |
|---|---|---|---|---|
| Immunomodulatory Drugs (IMiDs) | Thalidomide Lenalidomide | CRBN-dependent degradation of IKZF1/3, immune modulation, anti-angiogenic/inflammatory | CRBN-Ikaros axis mutations/transcriptional regulation, IKZF1/3 protection, upregulation of IL-6/STAT3 pathway | [8,9,10,11,12,13,14,15,16,17] |
| Pomalidomide | ||||
| Proteasome Inhibitors (PIs) | Bortezomib Ixazomib | Inhibit 26S proteasome though reversibly binding the PSMB5 subunit | Proteasome subunit mutations or upregulation, de-differentiation, alternate proteostasis pathways, increased drug efflux | [18,19,20,21,22,23,24] |
| Carfilzomib | Same as others, but binds irreversibly, also binds to PSMB2 | |||
| Monoclonal Antibodies | Daratumumab Isatuximab Elotuzumab | CDC, ADCC, ADCP, for CD38 antibodies, direct induction of cellular apoptosis | Decreased target expression, increased expression of complement inhibitory proteins | [25] |
| Glucocorticoids | Dexamethasone | Repress anti-apoptotic and metabolic pathways | Decreased GR expression, GR mutations | [26] |
| Chemotherapies | Melphalan | Causes DNA damage through alkylation | Increased drug efflux, increased expression of DNA repair factors | [27,28] |
| Current MM Drug Class | New Drug Class | Drug Name | Drug Targets | Clinical Trial Status (Clinical Trial #) | Refs |
|---|---|---|---|---|---|
| Immunomodulatory Drugs (IMiDs) | CELMoDs | Iberdomide (CC-220) Avadomide (CC-122) CC-92480 | Cereblon | Phase I/II (NCT02773030) Phase I (NCT01421524) | [52,53] |
| Phase I (NCT03374085) | |||||
| Proteasome Inhibitors (PIs) | PIs | Marizomib (NPI-0052) | β5, β2, and β1 subunits of 20S proteasome | Completed Phase II (NCT00461045) | [55] |
| Autophagy inhibitor | Hydroxychloroquine | Autolysosome formation | Completed Phase I (NCT00568880) | [56] | |
| Monoclonal Antibodies (mAbs) | CAR-T cells | Ide-Cel (bb2121) | BCMA | FDA approved for RRMM Phase III (NCT03361748) | [57,58] |
| Cilta-Cel (JNJ-4528) | Phase III (NCT04181827) | ||||
| BCMA-CART | Phase I (NCT02215967) | ||||
| Bispecific antibodies | REGN5458 | BCMAxCD3 | Phase I/II (NCT03761108) | [59,60,61] | |
| CC-93269 | Phase I (NCT03486067) | ||||
| Teclistamab | Phase II (NCT04557098) | ||||
| PF-3135 | Phase I (NCT03269136) | ||||
| AMG 701 | Phase I (NCT03287908) | ||||
| TNB-383B | Phase I (NCT03933735) | ||||
| Talquetamab | GPRC5DxCD3 | Phase I (NCT03399799) | |||
| Cevostamab | FCRH5xCD3 | Phase I (NCT03275103) | |||
| Antibody-drug conjugates | Belantamab mafodotin (GSK2857916) | BCMA | FDA approved for RRMM | [62,63] | |
| FOR46 | CD46 | Phase I (NCT03650491) | |||
| Other | SINE | Selinexor | XPO1 | FDA approved for RRMM | [64] |
| HDAC inhibitors | Ricolinostat | HDAC6 | Phase I/II (NCT01997840) | [65] | |
| Peptide-drug conjugates | Melflufen | Plasma cells (lipophilic cells) | FDA approved for RRMM Phase II (NCT02963493) | [66] | |
| BH3 mimetics | Venetoclax | BCL-2 | Phase III (NCT2755597); t (11;14) RRMM Phase I/II (NCT01794520) | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, L.N.; Sherbenou, D.W. Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma. Cancers 2021, 13, 1686. https://doi.org/10.3390/cancers13071686
Davis LN, Sherbenou DW. Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma. Cancers. 2021; 13(7):1686. https://doi.org/10.3390/cancers13071686
Chicago/Turabian StyleDavis, Lorraine N., and Daniel W. Sherbenou. 2021. "Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma" Cancers 13, no. 7: 1686. https://doi.org/10.3390/cancers13071686
APA StyleDavis, L. N., & Sherbenou, D. W. (2021). Emerging Therapeutic Strategies to Overcome Drug Resistance in Multiple Myeloma. Cancers, 13(7), 1686. https://doi.org/10.3390/cancers13071686