
PGC1α Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition


 , , and
, , and 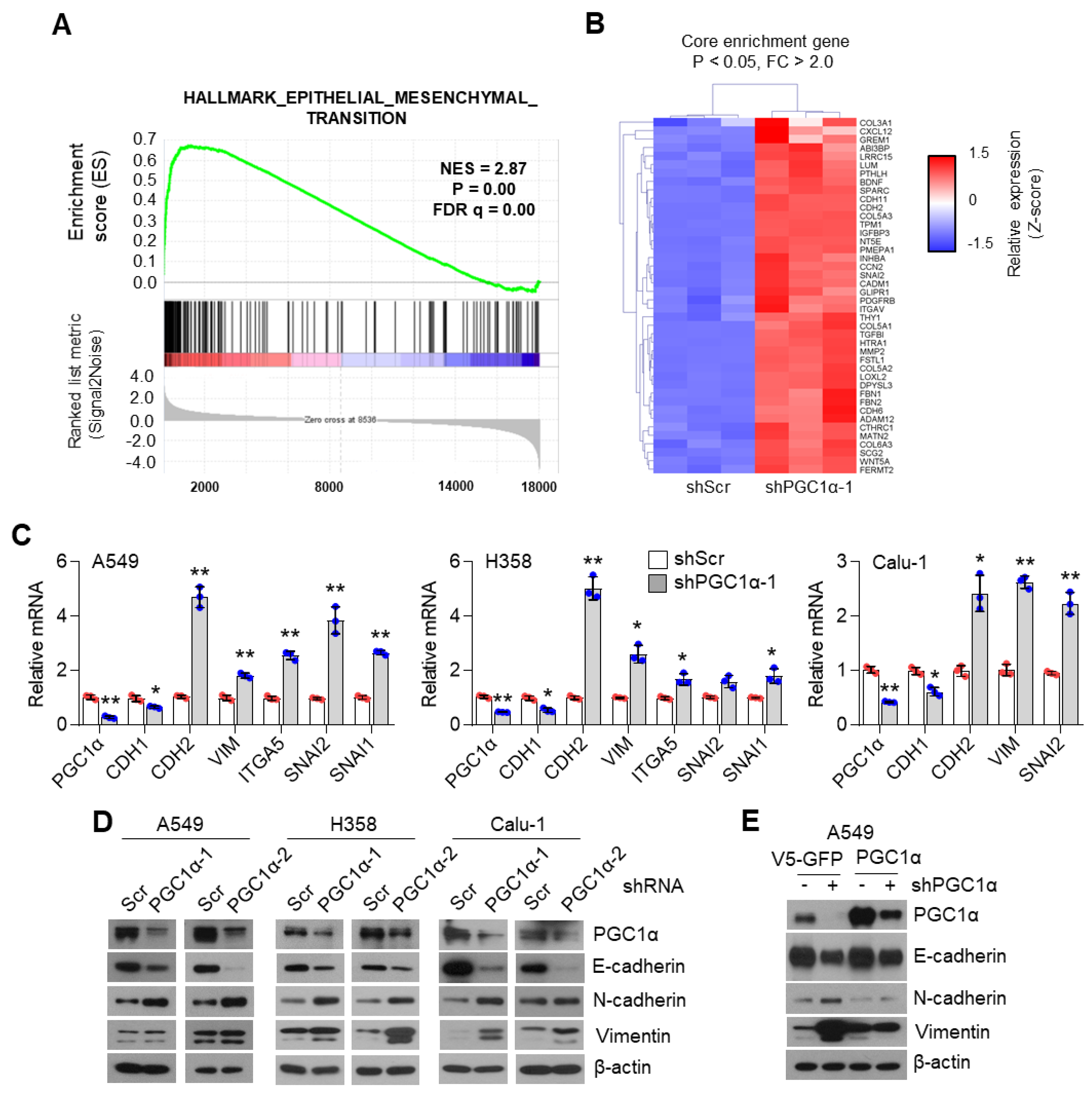{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-seq, GSEA, and Core Enrichment Gene Set Analysis
2.2. Bioinformatic Analysis Using Human Lung Cancer Biopsies
2.3. Human Lung Cancer Patient Samples and Immunohistochemistry
2.4. Tumor Xenograft in Mice and Serial Dilution In Vivo
2.5. Lung Orthotopic and Tail Vein Injection Xenograft Model
2.6. Kras Transgenic Mice
2.7. Generation of Ppargc1α (Pgc1α) knock-Out Mouse Using CRISPR-Cas9
2.8. FDG-Position Emission Tomography/Computed Tomography (PET/CT) Scanning
2.9. Cell Culture, Lentiviral Transduction and Generation of Stable Cell Lines
2.10. Quantitative Real-Time PCR (qRT-PCR) for Measurement of Gene Expression
2.11. Western Blotting and Co-Immunoprecipitation
2.12. In Vitro Migration and Invasion Assay
2.13. Transient Transfection
2.14. Chromatin Immunoprecipitation and Polymerase Chain Reaction (ChIP-PCR)
2.15. Luciferase Assay
2.16. Cell Viability and Annexin-V Assay
2.17. Statistical Analysis
3. Results
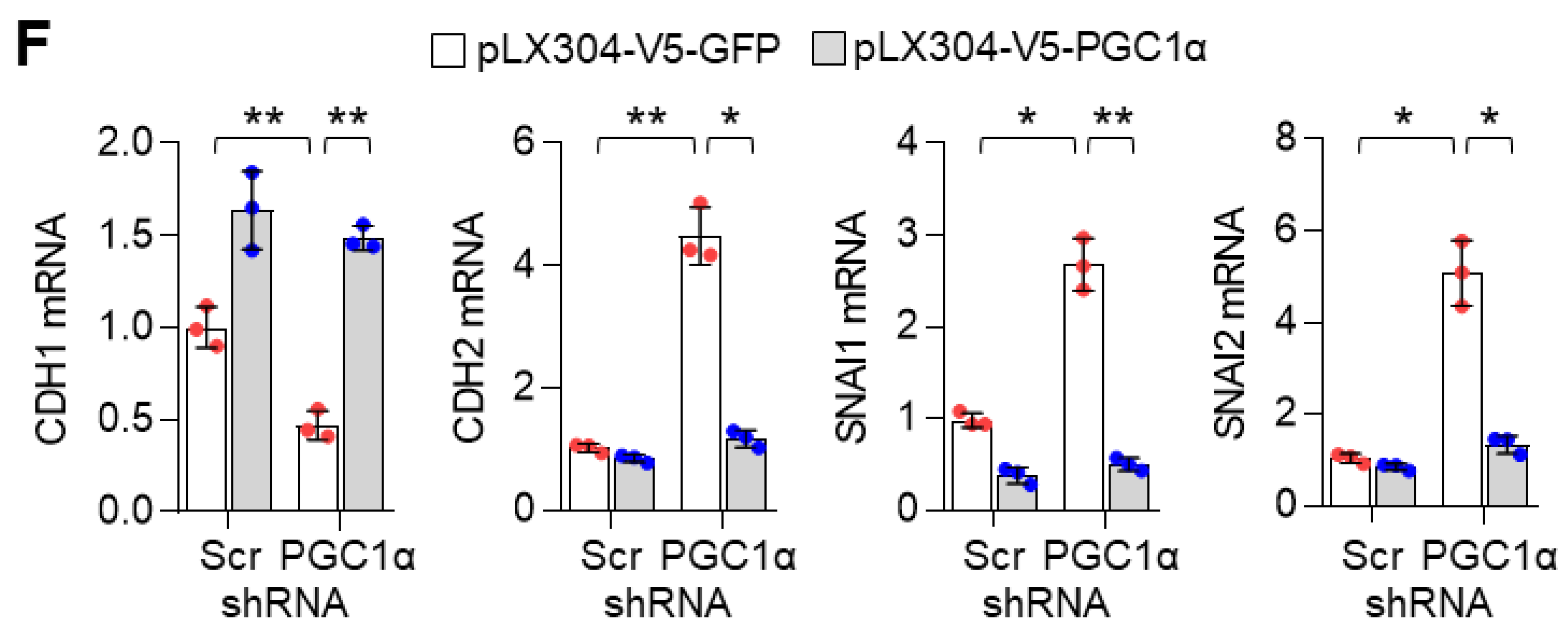
3.1. PGC1α Loss Promotes EMT in Lung Cancer Cells
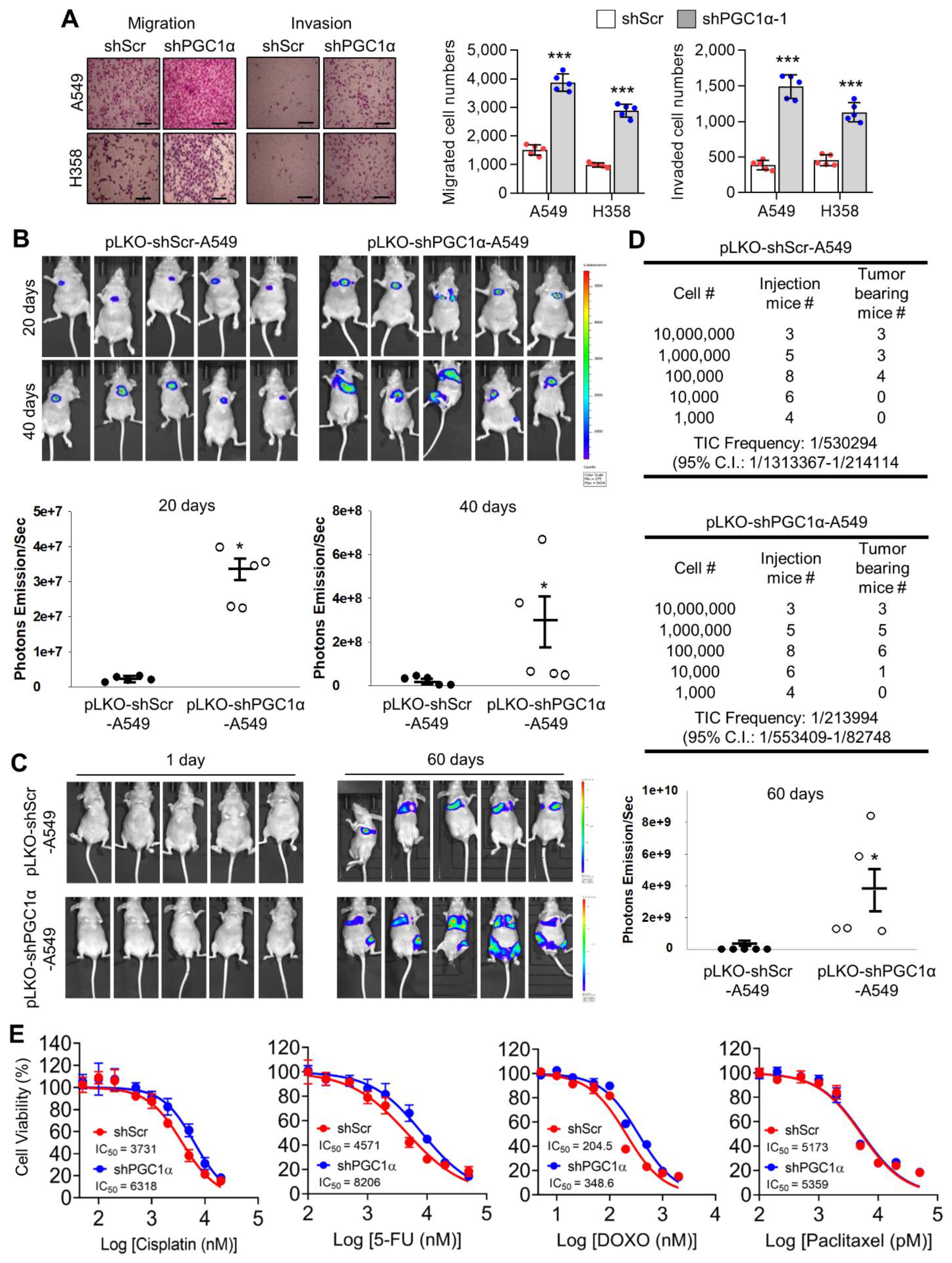
3.2. PGC1α Suppression Promotes Lung Cancer Initiation, Growth and Bone Metastasis
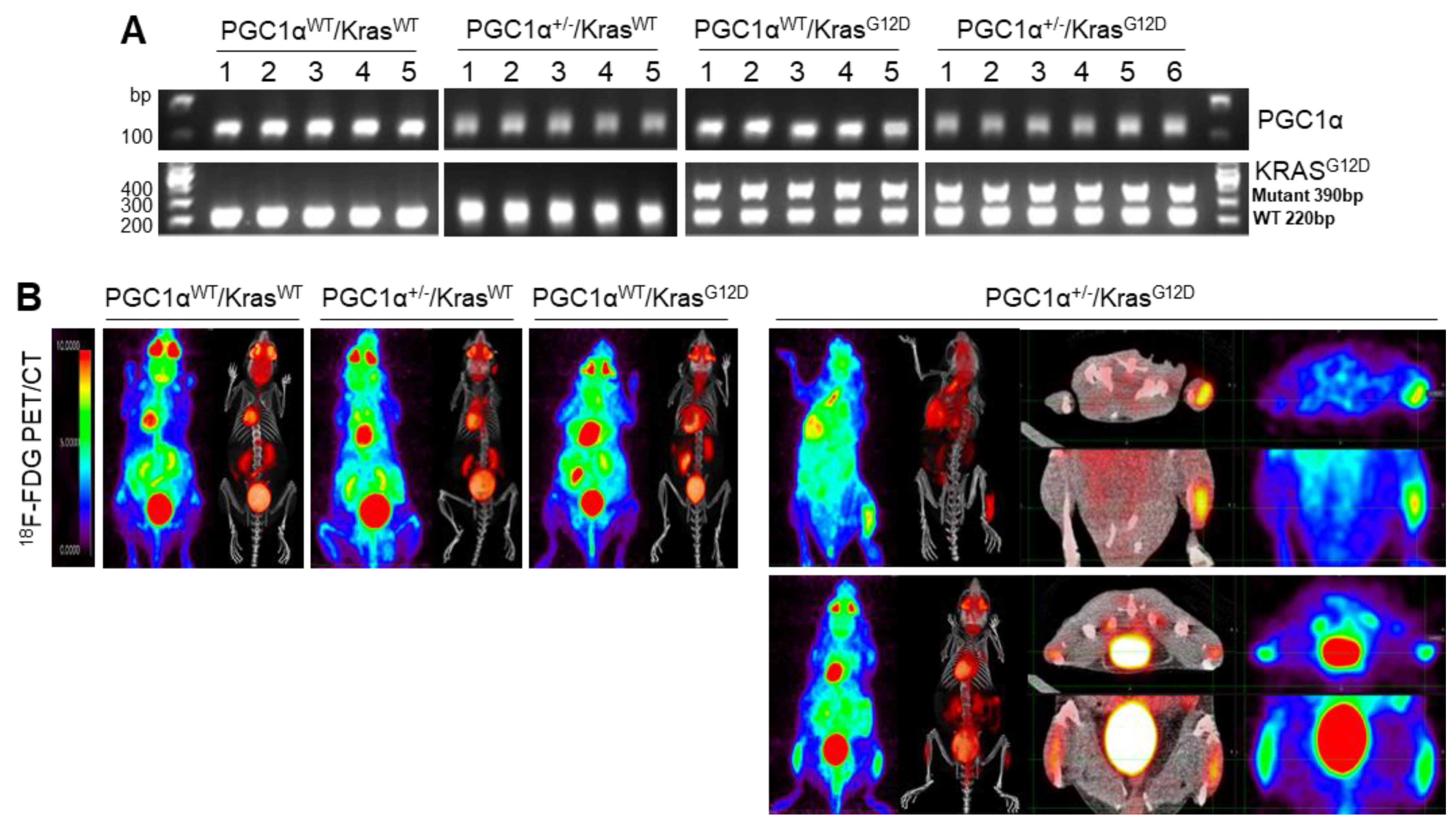
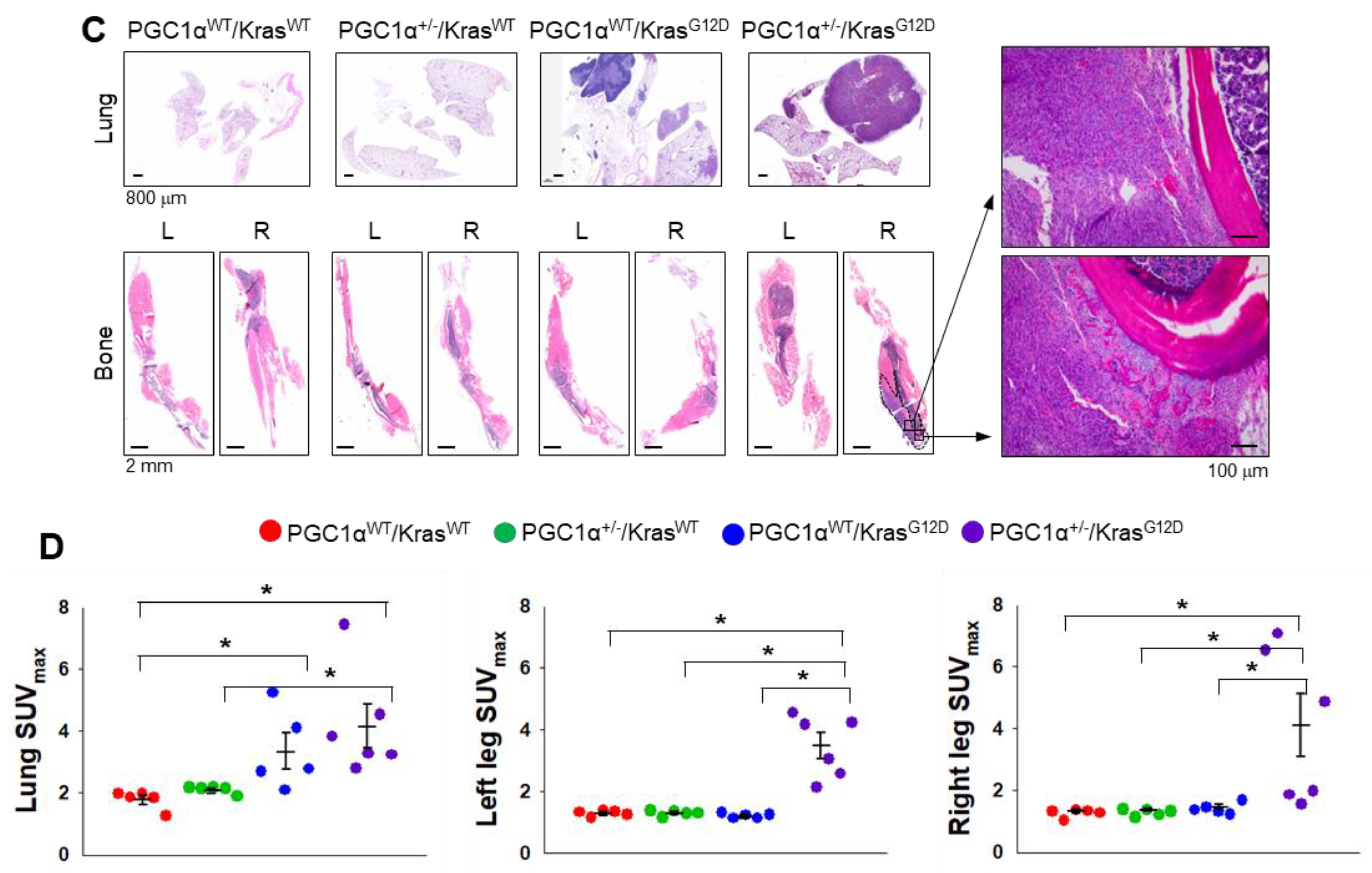
3.3. A Single Allele Deletion of Pgc1α Promotes Bone Metastasis of KrasG12D-Driven Lung Cancer
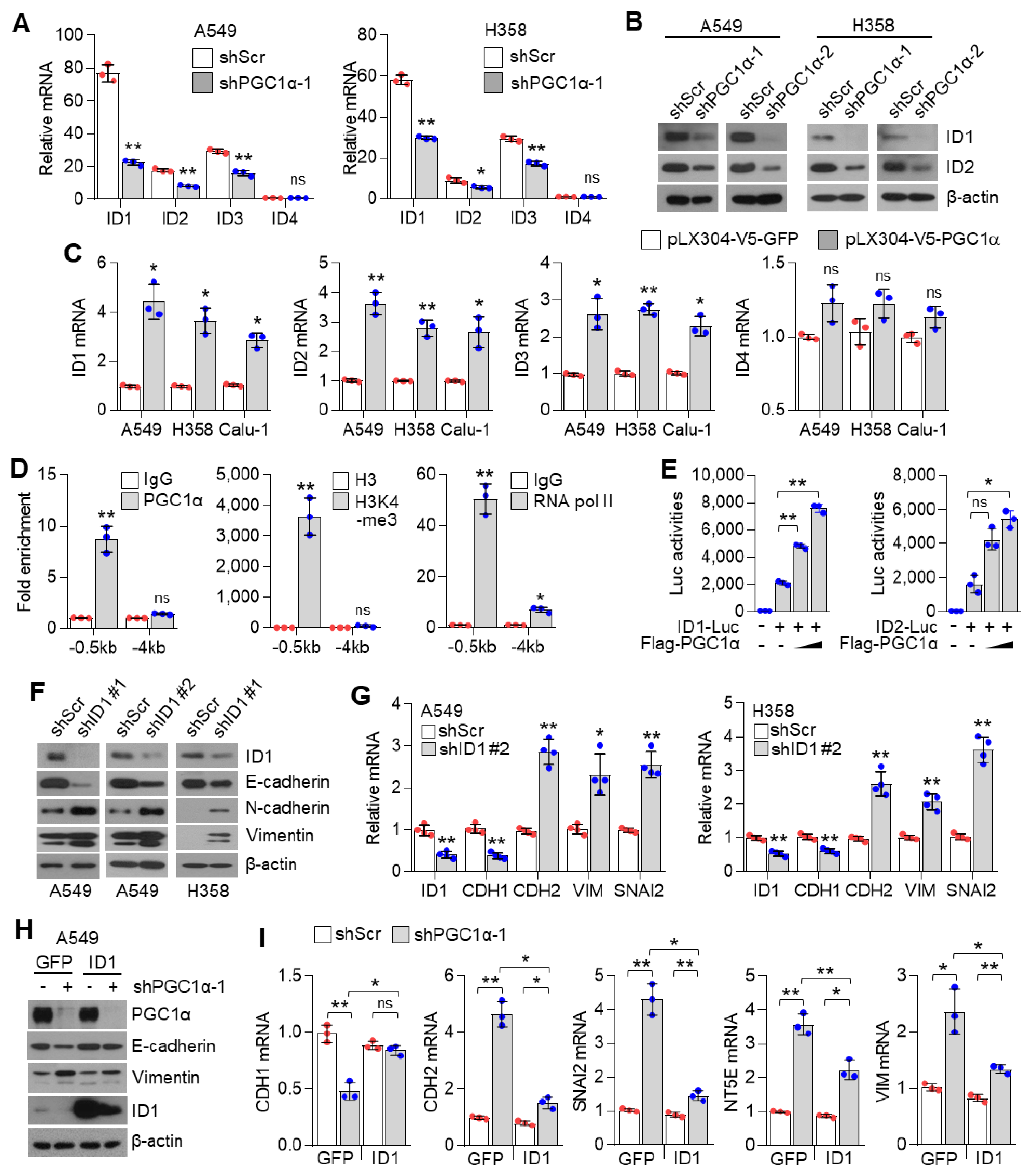
3.4. ID1 Is Required for the Loss of PGC1α-Mediated EMT
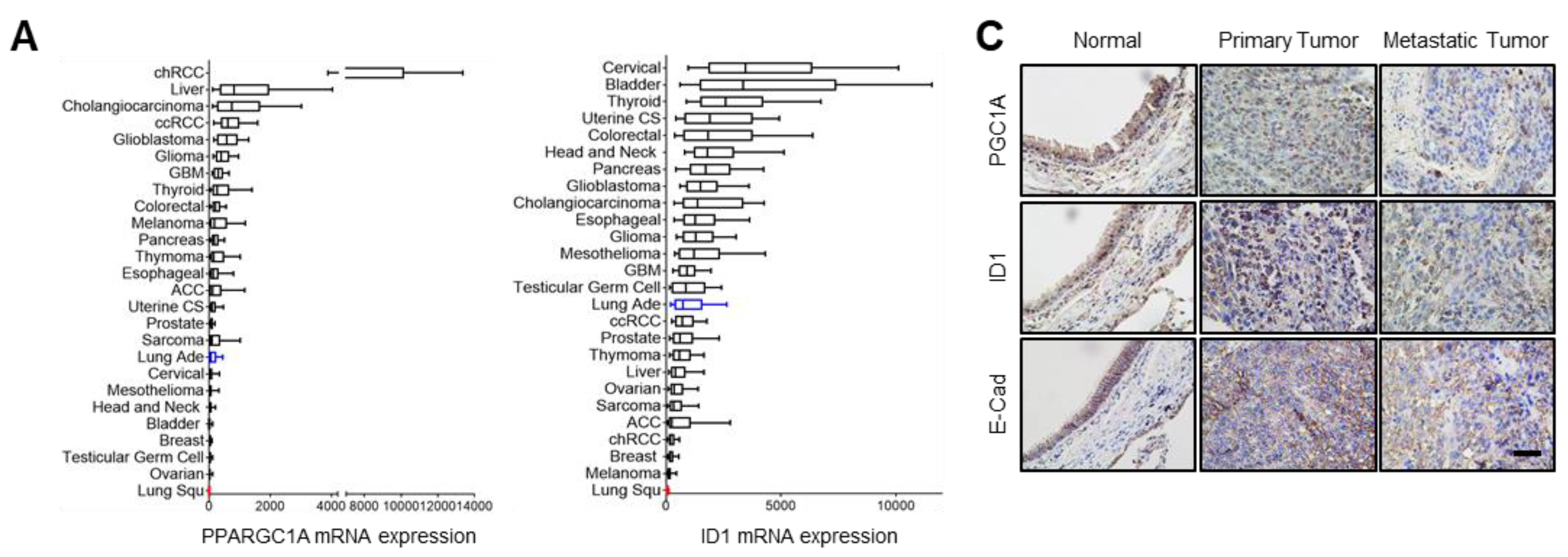
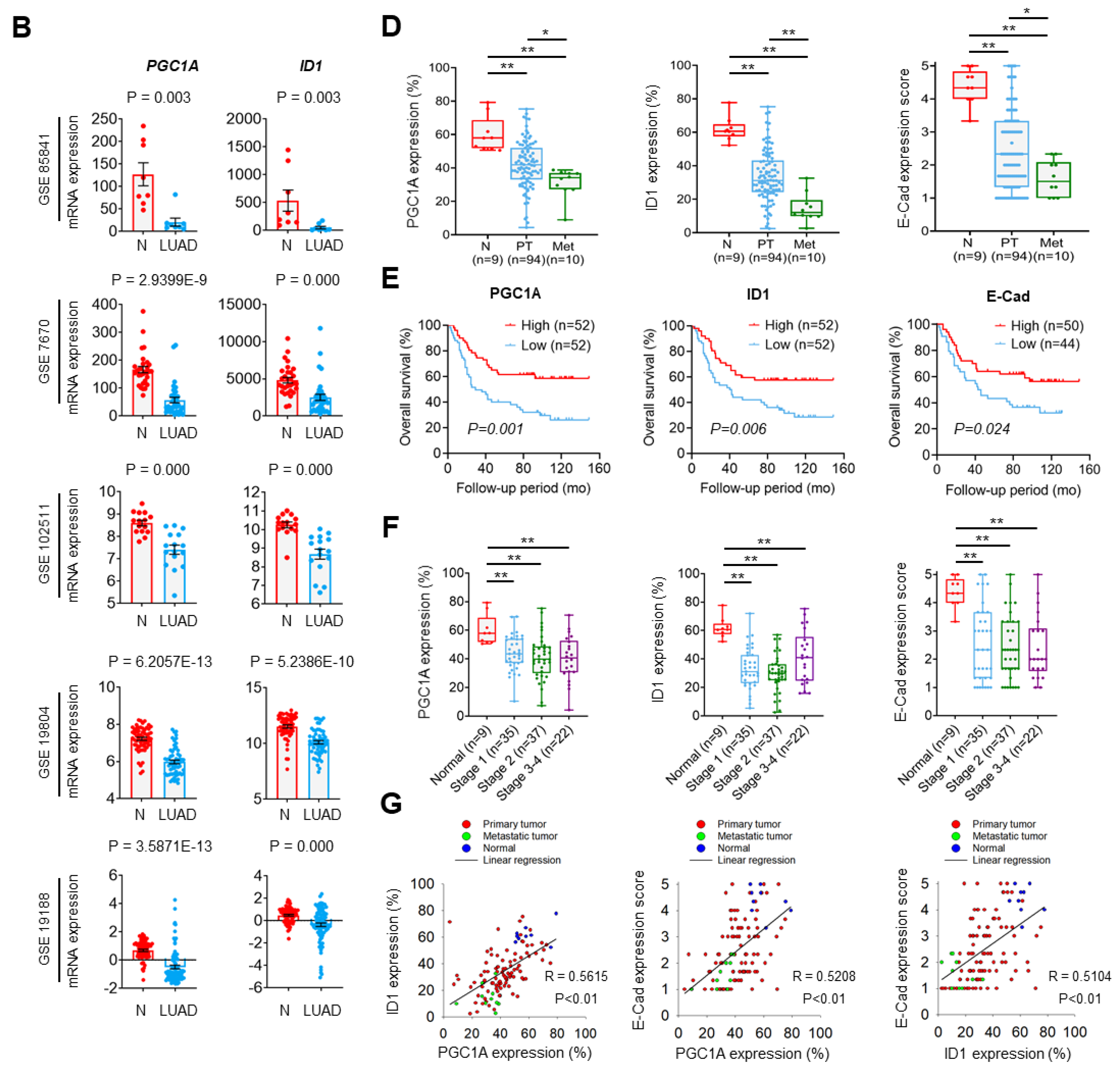
3.5. PGC1α and ID1 Is Decreased in Lung Cancer and Associated with a Poor Clinical Outcome
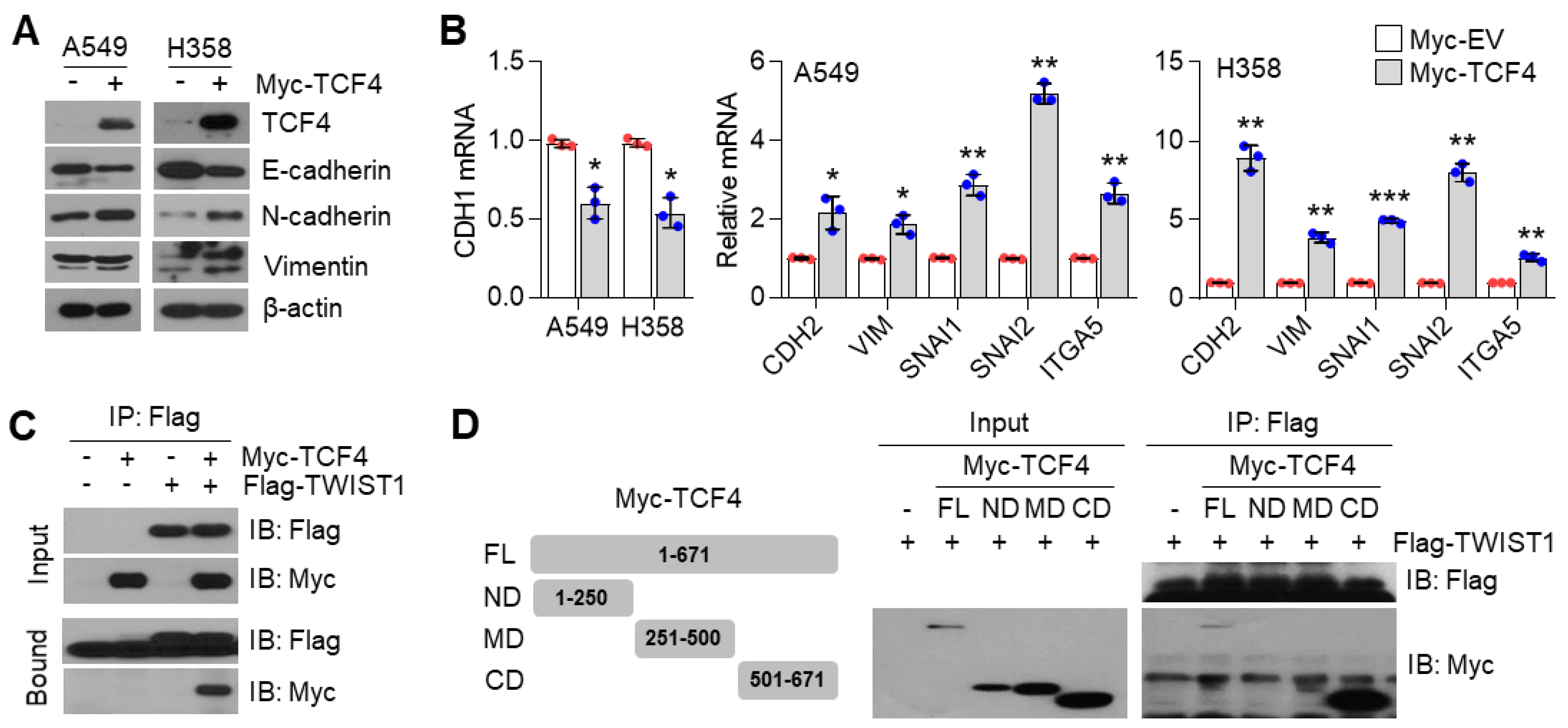
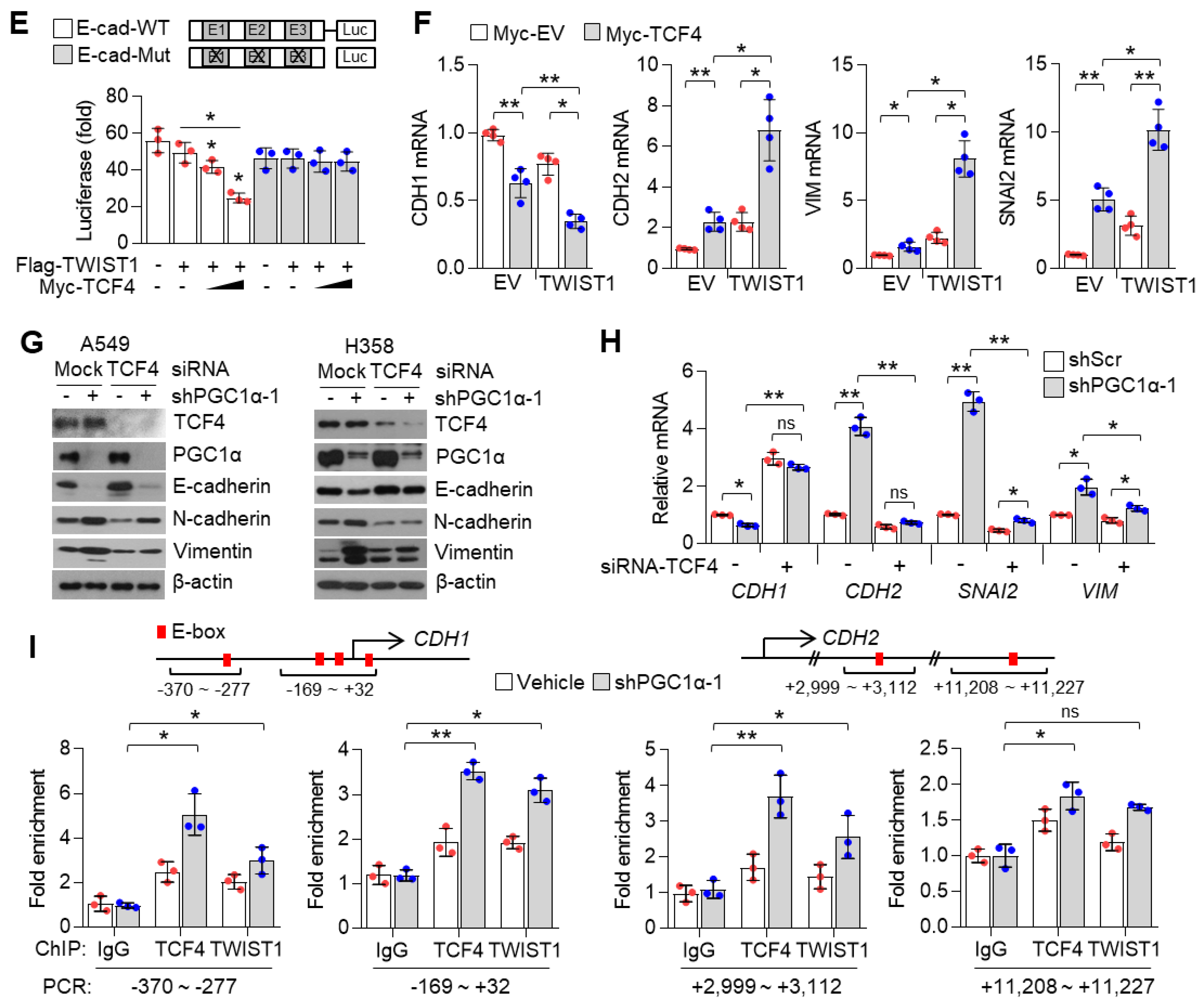
3.6. TCF4 Promotes TWIST1-Mediated EMT
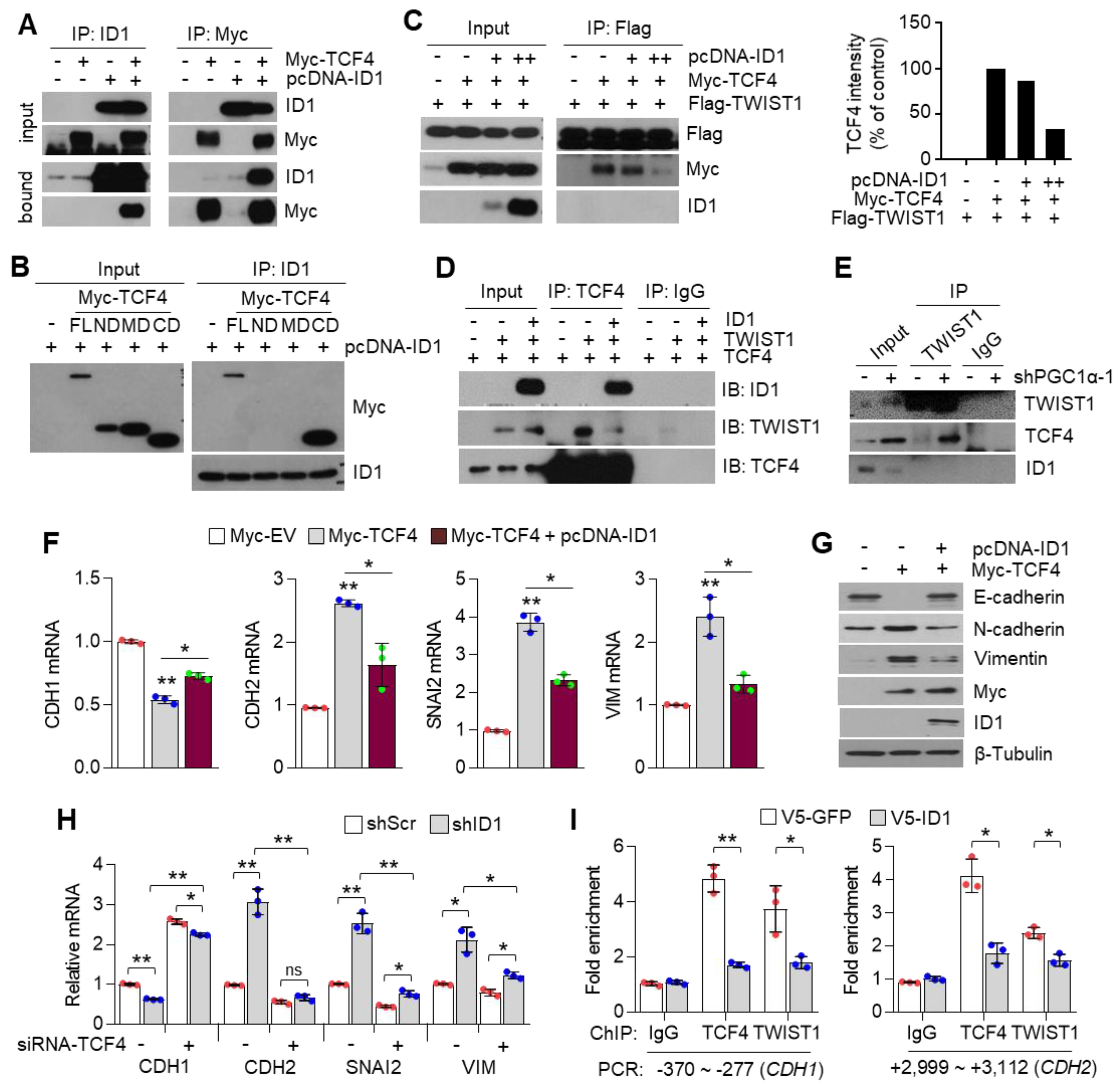
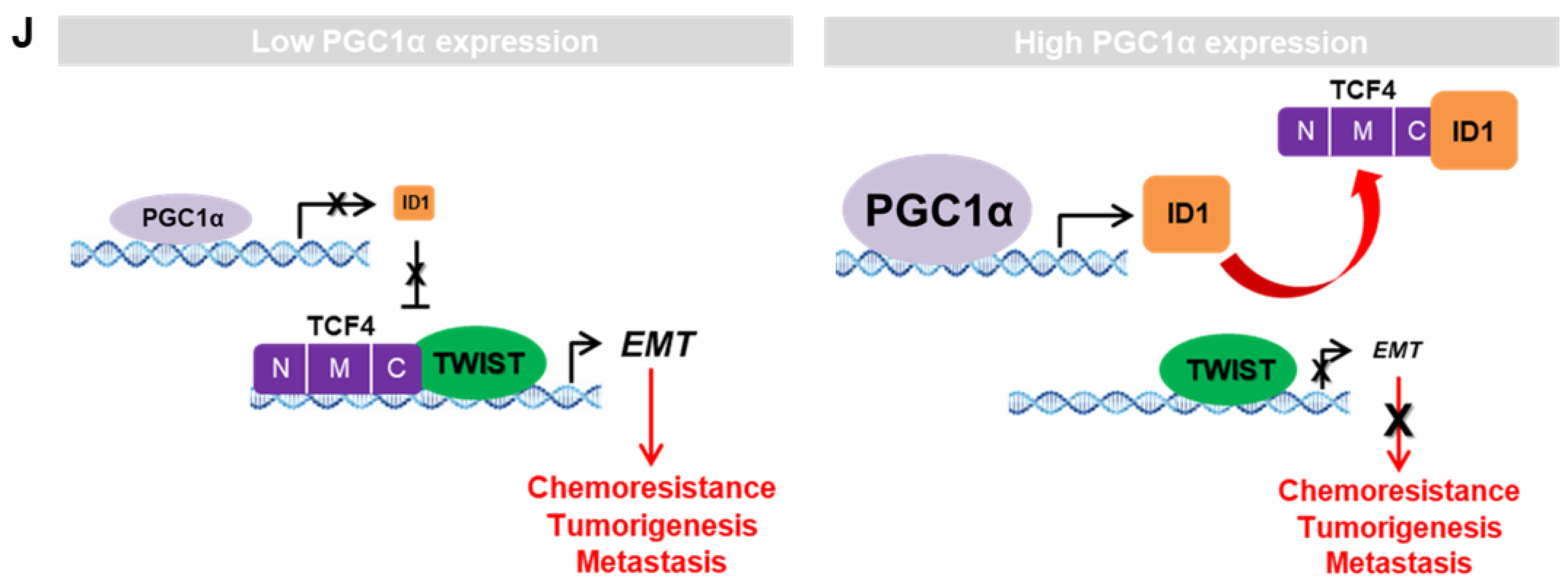
3.7. ID1 Attenuates EMT by Interfering with the TCF4-TWIST1 Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Antonio, C.; Passaro, A.; Gori, B.; Del Signore, E.; Migliorino, M.R.; Ricciardi, S.; Fulvi, A.; de Marinis, F. Bone and brain metastasis in lung cancer: Recent advances in therapeutic strategies. Ther. Adv. Med. Oncol. 2014, 6, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Gitelman, I. Twist protein in mouse embryogenesis. Dev. Biol. 1997, 189, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leptin, M. Twist and snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991, 5, 1568–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernandez-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Shoag, J.; Haq, R.; Zhang, M.; Liu, L.; Rowe, G.C.; Jiang, A.; Koulisis, N.; Farrel, C.; Amos, C.I.; Wei, Q.; et al. PGC-1 coactivators regulate MITF and the tanning response. Mol. Cell 2013, 49, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, G.; Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Choi, K.M.; Haak, A.J.; Aravamudhan, A.; Roden, A.C.; Prakash, Y.S.; et al. CBX5/G9a/H3K9me-mediated gene repression is essential to fibroblast activation during lung fibrosis. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1alpha repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Larson Casey, J.L.; Andrabi, S.A.; Lee, J.H.; Meza-Perez, S.; Randall, T.D.; Carter, A.B. Mitochondrial calcium uniporter regulates PGC-1alpha expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019, 26, 101307. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef]
- Cruz-Bermudez, A.; Vicente-Blanco, R.J.; Laza-Briviesca, R.; Garcia-Grande, A.; Laine-Menendez, S.; Gutierrez, L.; Calvo, V.; Romero, A.; Martin-Acosta, P.; Garcia, J.M.; et al. PGC-1alpha levels correlate with survival in patients with stage III NSCLC and may define a new biomarker to metabolism-targeted therapy. Sci. Rep. 2017, 7, 16661. [Google Scholar] [CrossRef] [Green Version]
- Sobrado, V.R.; Moreno-Bueno, G.; Cubillo, E.; Holt, L.J.; Nieto, M.A.; Portillo, F.; Cano, A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J. Cell Sci. 2009, 122, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, I.; Eriksson, M.; Saarikettu, J.; Eriksson, B.; Corneliussen, B.; Grundstrom, T.; Holmberg, D. The basic helix-loop-helix transcription factor E2-2 is involved in T lymphocyte development. Eur. J. Immunol. 2000, 30, 2857–2863. [Google Scholar] [CrossRef]
- Flora, A.; Garcia, J.J.; Thaller, C.; Zoghbi, H.Y. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 15382–15387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grill, J.I.; Herbst, A.; Brandl, L.; Kong, L.; Schneider, M.R.; Kirchner, T.; Wolf, E.; Kolligs, F.T. Inactivation of Itf2 promotes intestinal tumorigenesis in Apc(Min/+) mice. Biochem. Biophys. Res. Commun. 2015, 461, 249–253. [Google Scholar] [CrossRef]
- Herbst, A.; Bommer, G.T.; Kriegl, L.; Jung, A.; Behrens, A.; Csanadi, E.; Gerhard, M.; Bolz, C.; Riesenberg, R.; Zimmermann, W.; et al. ITF-2 is disrupted via allelic loss of chromosome 18q21, and ITF-2B expression is lost at the adenoma-carcinoma transition. Gastroenterology 2009, 137, 639–648.e631. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Choi, H.; So, D.; Kim, Y.I.; Cho, K.; Chung, H.J.; Lee, K.H.; Chun, Y.S.; Cho, C.H.; Kang, G.H.; et al. ITF2 prevents activation of the beta-catenin-TCF4 complex in colon cancer cells and levels decrease with tumor progression. Gastroenterology 2014, 147, 430–442.e438. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L.; Dekhil, H.; Ceccon, M.; Purgante, S.; Lan, C.; Cleris, L.; Magistroni, V.; Formelli, F.; Gambacorti-Passerini, C.B. Colorectal tumors are effectively eradicated by combined inhibition of {beta}-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Res. 2010, 70, 7253–7263. [Google Scholar] [CrossRef] [Green Version]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef]
- Roschger, C.; Cabrele, C. The Id-protein family in developmental and cancer-associated pathways. Cell Commun Signal. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikder, H.A.; Devlin, M.K.; Dunlap, S.; Ryu, B.; Alani, R.M. Id proteins in cell growth and tumorigenesis. Cancer Cell 2003, 3, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, Y.M.; Oh, T.I.; Shin, D.H.; Kim, G.H.; Kan, S.Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Vicent, S.; Luis-Ravelo, D.; Anton, I.; Garcia-Tunon, I.; Borras-Cuesta, F.; Dotor, J.; De Las Rivas, J.; Lecanda, F. A novel lung cancer signature mediates metastatic bone colonization by a dual mechanism. Cancer Res. 2008, 68, 2275–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, L.; Torrino, S.; Dufies, M.; Djabari, Z.; Haider, R.; Roustan, F.R.; Jaune, E.; Laurent, K.; Nottet, N.; Michiels, J.F.; et al. PGC1alpha Inhibits Polyamine Synthesis to Suppress Prostate Cancer Aggressiveness. Cancer Res. 2019, 79, 3268–3280. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef]
- Fong, S.; Itahana, Y.; Sumida, T.; Singh, J.; Coppe, J.P.; Liu, Y.; Richards, P.C.; Bennington, J.L.; Lee, N.M.; Debs, R.J.; et al. Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 13543–13548. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.I.; et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Kamalian, L.; Gosney, J.R.; Forootan, S.S.; Foster, C.S.; Bao, Z.Z.; Beesley, C.; Ke, Y. Increased expression of Id family proteins in small cell lung cancer and its prognostic significance. Clin. Cancer Res. 2008, 14, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Park, T.I.; Lee, Y.M.; Jo, Y.M.; Kim, S. Expression of Id-1 and VEGF in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2013, 6, 2102–2111. [Google Scholar]
- Pillai, S.; Rizwani, W.; Li, X.; Rawal, B.; Nair, S.; Schell, M.J.; Bepler, G.; Haura, E.; Coppola, D.; Chellappan, S. ID1 facilitates the growth and metastasis of non-small cell lung cancer in response to nicotinic acetylcholine receptor and epidermal growth factor receptor signaling. Mol. Cell Biol. 2011, 31, 3052–3067. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Cubillo, E.; Tobiume, K.; Shirakihara, T.; Fukuda, N.; Suzuki, H.; Shimizu, K.; Takehara, K.; Cano, A.; Saitoh, M.; et al. A role for Id in the regulation of TGF-beta-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 2004, 11, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Sharma, B.K.; Elattar, S.; Chang, J.; Kapil, S.; Yuan, J.; Satyanarayana, A. Id1 Promotes Obesity by Suppressing Brown Adipose Thermogenesis and White Adipose Browning. Diabetes 2017, 66, 1611–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Waardenberg, A.J.; Demuth, M.; Osteil, P.; Sun, J.Q.J.; Loebel, D.A.F.; Graham, M.; Tam, P.P.L.; Fossat, N. TWIST1 Homodimers and Heterodimers Orchestrate Lineage-Specific Differentiation. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef]
- Singh, S.; Gramolini, A.O. Characterization of sequences in human TWIST required for nuclear localization. BMC Cell Biol. 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.I.; Park, J.S.; Kim, D.H.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1alpha Suppresses the Activation of TGF-beta/Smad Signaling via Targeting TGFbetaRI Downregulation by let-7b/c Upregulation. Int. J. Mol. Sci. 2019, 20, 5084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.Y.; Brunicardi, F.C.; Lin, X. Smad3 mediates immediate early induction of Id1 by TGF-beta. Cell Res. 2009, 19, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Veerasamy, M.; Phanish, M.; Dockrell, M.E. Smad mediated regulation of inhibitor of DNA binding 2 and its role in phenotypic maintenance of human renal proximal tubule epithelial cells. PLoS ONE 2013, 8, e51842. [Google Scholar] [CrossRef] [Green Version]
- Kakonen, S.M.; Selander, K.S.; Chirgwin, J.M.; Yin, J.J.; Burns, S.; Rankin, W.A.; Grubbs, B.G.; Dallas, M.; Cui, Y.; Guise, T.A. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 2002, 277, 24571–24578. [Google Scholar] [CrossRef] [Green Version]
- Waning, D.L.; Guise, T.A. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin. Cancer Res. 2014, 20, 3071–3077. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Baker, N.E. E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease. Dev. Cell 2015, 35, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Meller, N.; Cutchins, A.; Deliri, H.; Slayton, R.P.; Oldham, S.N.; Kim, J.B.; Keller, S.R.; McNamara, C.A. The helix-loop-helix factors Id3 and E47 are novel regulators of adiponectin. Circ. Res. 2008, 103, 624–634. [Google Scholar] [CrossRef]
- Li, X.; Luo, Y.; Starremans, P.G.; McNamara, C.A.; Pei, Y.; Zhou, J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat. Cell Biol. 2005, 7, 1202–1212. [Google Scholar] [CrossRef]
- Peddada, S.; Yasui, D.H.; LaSalle, J.M. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 2006, 15, 2003–2014. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Clawson, G.A.; Olivieri, N.F.; Bell, L.L.; Begley, C.G.; Miller, B.A. Expression of SCL is normal in transfusion-dependent Diamond-Blackfan anemia but other bHLH proteins are deficient. Blood 1997, 90, 2068–2074. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, T.-I.; Lee, M.; Lee, Y.-M.; Kim, G.-H.; Lee, D.; You, J.S.; Kim, S.H.; Choi, M.; Jang, H.; Park, Y.-M.; et al. PGC1α Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers 2021, 13, 1772. https://doi.org/10.3390/cancers13081772
Oh T-I, Lee M, Lee Y-M, Kim G-H, Lee D, You JS, Kim SH, Choi M, Jang H, Park Y-M, et al. PGC1α Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers. 2021; 13(8):1772. https://doi.org/10.3390/cancers13081772
Chicago/Turabian StyleOh, Taek-In, Mingyu Lee, Yoon-Mi Lee, Geon-Hee Kim, Daekee Lee, Jueng Soo You, Sun Ha Kim, Minyoung Choi, Hyonchol Jang, Yeong-Min Park, and et al. 2021. "PGC1α Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition" Cancers 13, no. 8: 1772. https://doi.org/10.3390/cancers13081772
APA StyleOh, T. -I., Lee, M., Lee, Y. -M., Kim, G. -H., Lee, D., You, J. S., Kim, S. H., Choi, M., Jang, H., Park, Y. -M., Shin, H. -W., Shin, D. H., & Lim, J. -H. (2021). PGC1α Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers, 13(8), 1772. https://doi.org/10.3390/cancers13081772