
Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution



Abstract
:Simple Summary
Abstract
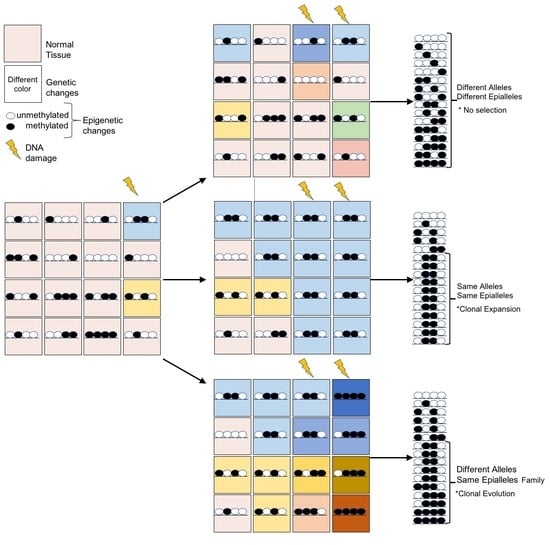
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cancer Evolution
3. Genome Instability (Genome Chaos)
4. Epigenome Chaos
5. Mechanisms of de Novo DNA Methylation
6. New Tools for DNA Methylation Analysis
7. Epialleles-Based Analysis (EBA)
- AmpliMethProfiler, a python-based pipeline, extracts and performs statistical epihaplotype analysis of amplicons from targeted deep bisulfite sequences. This tool investigates the methylation diversity by directly extracting the methylation profiles (epihaplotypes) at a single locus in the sequence population [84,86]. Using this high-throughput approach, the epihaplotypes can be treated as haploid organisms with a specific frequency in the population (Shannon Entropy).
- MethCoresProfiler, a R-based pipeline, traces and tracks CpGs in the same phase (methylated or not methylated cores) shared by families of epialleles by calculating their frequency in the population (MethCore Index), the frequency normalized to the mean methylation (Clonality Index), and the association index between the CpGs belonging to the same core normalized to the average methylation of the population of sequences (Entanglement Index) [83]. This tool is able to recognize the original epigenetic ancestor from which the molecules of different epialleles derive, considering each addition or removal of a methyl groups as independent events. This method allows the reconstruction of the evolution of families of epialleles from a common ancestor. Note that the frequency of individual epialleles is usually not statistically significant, while the frequency of the common signature (core) is significant. This tool analyses amplicons from targeted deep bisulfite sequencing and allows the analysis of several samples longitudinally.
- Methclone extracts and performs statistical epihaplotype analysis for each locus from genome-wide DNA NGS data (RRBS and WGBS). It is based on the comparison between two samples longitudinally and identifies the epigenetic loci hosting large clonal variations. It quantifies epiallele shift(s), as the Hamming distance and the frequency of single epialleles [85].
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APAF-1 | apoptotic protease activating factor 1 |
| ASXL | additional sex combs-like |
| ATM | ATM serine/threonine kinase or ataxia-telangiectasia mutated |
| BRCA1 | breast cancer type 1 |
| C | cytosine |
| CDKN2A | Cyclin Dependent Kinase Inhibitor 2A |
| CDKN2B | Cyclin Dependent Kinase Inhibitor 2B |
| CDK CpG | cyclin-dependent kinase cytosine-guanine dinucleotides cluster |
| DNMT1 | DNA methyl-transferase 1 |
| DNMT3a | DNA methyl-transferase 3A |
| DAPK | Death Associated Protein Kinase |
| DMC | differentially methylated cytosines |
| DMR | differentially methylated regions |
| DSB | double strand break |
| EBA | Epialleles-Based Analysis |
| GFP | green fluorescence protein |
| HR | homologous repair |
| hMLH1 | MutL (E. Coli) Homolog 1 |
| INK4 | INhibitors of CDK4-CDK6 |
| ARF | alternate open reading frame |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LOI | loss of imprinting |
| MGMT | O6-methylguanine-DNA methyltransferase |
| ROS | reactive oxygen species |
| RRBS | reduced representation bisulfite sequencing |
| TAD | topologically associating domains |
| TGF-β1 | Transforming growth factor beta1 |
| TAD | topologically associating domains |
| TSS | transcription start site |
| TET2 | Ten-Eleven Translocation |
| WGBS | whole-genome bisulfite sequencing |
| WIF1 | WNT Inhibitory Factor 1 |
| ZNF304 | Zinc Finger Protein 304 |
References
- Du, H.; Che, G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett. 2017, 13, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Grønbaek, K.; Hother, C.; Jones, P.A. Epigenetic changes in cancer. APMIS 2007, 115, 1039–1059. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer. 2013, 108, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 1–19. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Cuozzo, C.; Porcellini, A.; Angrisano, T.; Morano, A.; Lee, B.; Pardo, A.D.; Messina, S.; Iuliano, R.; Fusco, A.; Santillo, M.R.; et al. Correction: DNA damage, homology-directed repair, and dna methylation. PLoS Genet. 2017, 13, e1006605. [Google Scholar] [CrossRef]
- Morano, A.; Angrisano, T.; Russo, G.; Landi, R.; Pezone, A.; Bartollino, S.; Zuchegna, C.; Babbio, F.; Bonapace, I.M.; Allen, B.; et al. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 2014, 42, 804–821. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.; Landi, R.; Pezone, A.; Morano, A.; Zuchegna, C.; Romano, A.; Muller, M.T.; Gottesman, M.E.; Porcellini, A.; Accedimento, E.V. DNA damage and repair modify DNA methylation and chromatin domain of the targeted locus: Mechanism of allele methylation polymorphism. Sci. Rep. 2016, 6, 33222. [Google Scholar] [CrossRef] [Green Version]
- Alcin, M. The Runge Kutta-4 based 4D hyperchaotic system design for secure communication applications. Chaos Theory Appl. 2020, 2, 23–30. [Google Scholar]
- Podlaha, O.; Riester, M.; De, S.; Michor, F. Evolution of the cancer genome. Trends Genet. 2012, 28, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Tramontano, A.; Boffo, F.L.; Russo, G.; De Rosa, M.; Iodice, I.; Pezone, A. Methylation of the suppressor gene p16INK4a: Mechanism and consequences. Biomolecules 2020, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Tramontano, A.; Pezone, A.; Migliaccio, A. LSD1: More than demethylation of histone lysine residues. Exp. Mol. Med. 2020, 52, 1936–1947. [Google Scholar] [CrossRef]
- Li, S.; Kennedy, M.; Payne, S.; Kennedy, K.; Seewaldt, V.L.; Pizzo, S.V.; Bachelder, R.E. Model of tumor dormancy/recurrence after short-term chemotherapy. PLoS ONE 2014, 9, e98021. [Google Scholar] [CrossRef] [Green Version]
- De Pasquale, V.; Pezone, A.; Sarogni, P.; Tramontano, A.; Schiattarella, G.G.; Avvedimento, V.E.; Paladino, S.; Pavone, L.M. EGFR activation triggers cellular hypertrophy and lysosomal disease in NAGLU-depleted cardiomyoblasts, mimicking the hallmarks of mucopolysaccharidosis IIIB. Cell Death Dis. 2018, 9, 40. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020, S1044-579X, 30224–30228. [Google Scholar] [CrossRef]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.; Ye, K.; Bremer, S.; Ye, C.; Chen, D.J.; Heng, H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, H.H.; Stevens, J.B.; Bremer, S.W.; Liu, G.; Abdallah, B.Y.; Ye, C.J. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res. 2011, 112, 217–253. [Google Scholar]
- Stevens, J.B.; Liu, G.; Abdallah, B.Y.; Horne, S.D.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Krawetz, S.A.; Heng, H.H. Unstable genomes elevate transcriptome dynamics. Int. J. Cancer 2014, 134, 2074–2087. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Bremer, S.W.; Ye, K.J.; Abdallah, B.Y.; Horne, S.D.; Ye, C.J. Decoding the genome beyond sequencing: The new phase of genomic research. Genomics 2011, 98, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; Macdonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Karen, J.Y.; Christine, J.Y. Chromosomal instability [CIN]: What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef]
- Abdallah, B.Y.; Horne, S.D.; Stevens, J.B.; Liu, G.; Ying, A.Y.; Vanderhynden, B.; Krawetz, S.A.; Gorelick, R.; Heng, H.H.Q. Single cell heterogeneity: Why unstable genomes are incompatible with average profiles. Cell Cycle 2013, 12, 3640–3649. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H. The genome-centric concept: Resynthesis of evolutionary theory. Bioessays 2009, 31, 512–525. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritgenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Zhang, Y.; Risques, R.A. Cancer-associated mutations but no cancer: Insights into the early steps of carcinogenesis and implications for early cancer detection. Trends Cancer 2019, 5, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Gisselsson, D.; Dumanski, L.A.F.J.P. Mosaicism in health and disease—Clones picking up speed. Nat. Rev. Genet. 2017, 18, 128–142. [Google Scholar] [CrossRef]
- Krimmel, J.D.; Salk, J.J.; Risques, R.A. Cancer-like mutations in non-cancer tissue: Towards a better understanding of multistep carcinogenesis. Transl. Cancer Res. 2016, 5, S1302–S1304. [Google Scholar] [CrossRef]
- Franklin, W.A.; Gazdar, A.F.; Haney, J.; Wistuba, I.I.; La Rosa, F.G.; Kennedy, T.; Ritchey, D.M.; Miller, Y.E. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J. Clin. Investig. 1997, 100, 2133–2137. [Google Scholar] [CrossRef] [Green Version]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef] [Green Version]
- Sardo, V.L.; Ferguson, W.; Erikson, G.A.; Topol, E.J.; Baldwin, K.K.; Torkamani, A. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 2017, 35, 69–74. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.T.; Salk, J.J.; Brentnall, T.A.; Risques, R.A. Precancer in ulcerative colitis: The role of the field effect and its clinical implications. Carcinogenesis 2017, 39, 11–20. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nat. Cell Biol. 2019, 565, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nat. Cell Biol. 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Keogh, M.J.; Wei, W.; Aryaman, J.; Walker, L.; Ameele, J.V.D.; Coxhead, J.; Wilson, I.; Bashton, M.; Beck, J.; West, J.; et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Anglesio, M.S.; Papadoupoulos, N.; Ayhan, A.; Nazeran, T.M.; Noe, M.; Horlings, H.M.; Kum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med. 2018, 376, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nat. Cell Biol. 2020, 580, 640–646. [Google Scholar] [CrossRef]
- Salk, J.J.; Loubet-Senear, K.; Maritschnegg, E.; Valentine, C.C.; Williams, L.N.; Higgins, J.E.; Horvat, R.; Vanderstichele, A.; Nachmanson, D.; Baker, K.T.; et al. Ultra-sensitive TP53 sequencing for cancer detection reveals progressive clonal selection in normal tissue over a century of human lifespan. Cell Rep. 2019, 28, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Braakhuis, B.J.M.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.A.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef]
- Nandi, B.; Talluri, S.; Kumar, S.; Yenumula, S.; Gold, J.S.; Prabhala, R.; Munshi, N.C.; Shammas, M.A. The roles of homologous recombination and the immune system in the genomic evolution of cancer. J. Transl. Sci. 2019, 5. [Google Scholar] [CrossRef]
- Heuser, M.; Thol, F.; Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch. Aerzteblatt Online 2016, 113, 317–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Angrisano, T.; Pero, R.; Brancaccio, M.; Coretti, L.; Florio, E.; Pezone, A.; Calabrò, V.; Falco, G.; Keller, S.; Lembo, F.; et al. Cyclical DNA methylation and histone changes are induced by lps to activate cox-2 in human intestinal epithelial cells. PLoS ONE 2016, 11, e0156671. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.-H.; Lee, T.-L.; Rennert, O.M.; Chan, W.-Y. DNA methylation of cancer genome. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-W.; Idrees, K.; Shattock, R.; Khan, S.A.; Zeng, Z.; Brennan, C.W.; Paty, P.; Barany, F. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int. J. Cancer 2010, 127, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Putiri, E.L.; Robertson, K.D. Epigenetic mechanisms and genome stability. Clin. Epigenetics 2010, 2, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Huang, Y.-H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Pezone, A.; Russo, G.; Tramontano, A.; Florio, E.; Scala, G.; Landi, R.; Zuchegna, C.; Romano, A.; Chiariotti, L.; Muller, M.T.; et al. High-coverage methylation data of a gene model before and after DNA damage and homologous repair. Sci. Data 2017, 4, 170043. [Google Scholar] [CrossRef] [Green Version]
- Pezone, A.; Zuchegna, C.; Tramontano, A.; Romano, A.; Russo, G.; De Rosa, M.; Vinciguerra, M.; Porcellini, A.; Gottesman, M.E.; Avvedimento, E.V. RNA stabilizes transcription-dependent chromatin loops induced by nuclear hormones. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change. Oncotarget 2017, 8, 40359–40372. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Serra, R.W.; Fang, M.; Park, S.M.; Hutchinson, L.; Green, M.R. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. eLife 2014, 3, e02313. [Google Scholar] [CrossRef]
- Svegliati, S.; Marrone, G.; Pezone, A.; Spadoni, T.; Grieco, A.; Moroncini, G.; Grieco, D.; Vinciguerra, M.; Agnese, S.; Jüngel, A.; et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the Wnt inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci. Signal. 2014, 7, ra84. [Google Scholar] [CrossRef]
- Pezone, A.; Taddei, M.L.; Tramontano, A.; Dolcini, J.; Boffo, F.L.; De Rosa, M.; Parri, M.; Stinziani, S.; Comito, G.; Porcellini, A.; et al. Targeted DNA oxidation by LSD1–SMAD2/3 primes TGF-β1/ EMT genes for activation or repression. Nucleic Acids Res. 2020, 48, 8943–8958. [Google Scholar] [CrossRef]
- Weaver, J.R.; Susiarjo, M.; Bartolomei, M.S. Imprinting and epigenetic changes in the early embryo. Mamm. Genome 2009, 20, 532–543. [Google Scholar] [CrossRef]
- Bashtrykov, P.; Jankevicius, G.; Smarandache, A.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 2012, 19, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.A.; Aranda, V.V.; Bardelli, A.A.; Blanpain, C.; Bock, C.C.; Borowski, C.C.; Caldas, C.; Califano, A.A.; Doherty, M.M.; Elsner, M.M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Mikeska, T.; Candiloro, I.L.M.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Pezone, A.; Tramontano, A.; Scala, G.; Cuomo, M.; Riccio, P.; De Nicola, S.; Porcellini, A.; Chiariotti, L.; Avvedimento, E.V. Tracing and tracking epiallele families in complex DNA populations. NAR Genom. Bioinform. 2020, 2, lqaa096. [Google Scholar] [CrossRef]
- Scala, G.; Affinito, O.; Palumbo, D.; Florio, E.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. ampliMethProfiler: A pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons. BMC Bioinform. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Garrett-Bakelman, F.; Perl, A.E.; Luger, S.M.; Zhang, C.; To, B.L.; Lewis, I.D.; Brown, A.L.; D’Andrea, R.J.; Ross, M.E.; et al. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Cuomo, M.; Keller, S.; Punzo, D.; Nuzzo, T.; Affinito, O.; Coretti, L.; Carella, M.; De Rosa, V.; Florio, E.; Boscia, F.; et al. Selective demethylation of two CpG sites causes postnatal activation of the Dao gene and consequent removal of d-serine within the mouse cerebellum. Clin. Epigenetics 2019, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.-J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, G.; Tramontano, A.; Iodice, I.; Chiariotti, L.; Pezone, A. Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution. Cancers 2021, 13, 1800. https://doi.org/10.3390/cancers13081800
Russo G, Tramontano A, Iodice I, Chiariotti L, Pezone A. Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution. Cancers. 2021; 13(8):1800. https://doi.org/10.3390/cancers13081800
Chicago/Turabian StyleRusso, Giusi, Alfonso Tramontano, Ilaria Iodice, Lorenzo Chiariotti, and Antonio Pezone. 2021. "Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution" Cancers 13, no. 8: 1800. https://doi.org/10.3390/cancers13081800
APA StyleRusso, G., Tramontano, A., Iodice, I., Chiariotti, L., & Pezone, A. (2021). Epigenome Chaos: Stochastic and Deterministic DNA Methylation Events Drive Cancer Evolution. Cancers, 13(8), 1800. https://doi.org/10.3390/cancers13081800