
Myelodysplasia Syndrome, Clonal Hematopoiesis and Cardiovascular Disease

 , ,
, ,
Abstract
:Simple Summary
Abstract
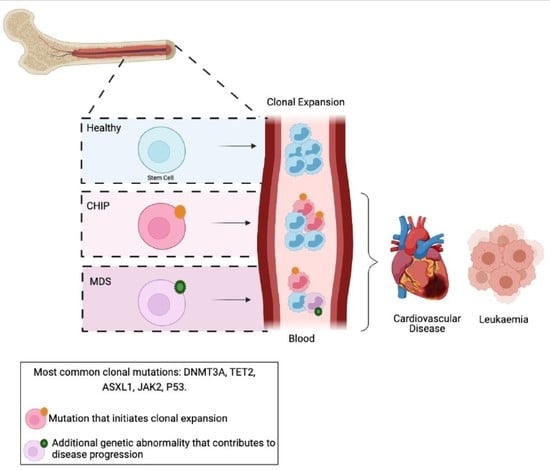
1. Introduction
2. MDS Pathophysiology
2.1. MDS Precursor Conditions
2.1.1. CHIP
2.1.2. CCUS and ICUS
3. Genetic Abnormalities in MDS Development
3.1. Somatic Mutations
3.1.1. Epigenetic Regulators
3.1.2. RNA Spliceosome
3.1.3. DNA Transcription
3.1.4. Other Genes
3.2. Germline Mutations
3.3. Chromosomal Abnormalities
3.4. MDS Progression to AML
4. The Dysfunction of CHIP-Related Genes Alters HSC Function
4.1. DNMTs
4.2. TET2
DNMT3A and TET2 Co-Deletion
4.3. P53
4.4. ASXL1
4.5. JAK2
5. CHIP—A Novel Connection between CVD and MDS
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Hasserjian, R.P. Myelodysplastic Syndrome Updated. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2019, 86, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Blonquist, T.M.; Hobbs, G.S.; Amrein, P.C.; Neuberg, D.S.; Steensma, D.P.; Abel, G.A.; Fathi, A.T. Risk and timing of cardiovascular death among patients with myelodysplastic syndromes. Blood Adv. 2017, 1, 2032–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P. Graphical representation of clinical outcomes for patients with myelodysplastic syndromes. Leuk. Lymphoma 2016, 57, 17–20. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Hasserjian, R.P. Controversies in the recent (2016) World Health Organization classification of acute myeloid leukemia. Best Pr. Res. Clin. Haematol. 2021, 34, 101249. [Google Scholar] [CrossRef]
- Andersen, M.K.; Christiansen, D.H.; Pedersen-Bjergaard, J. Centromeric breakage and highly rearranged chromosome derivatives associated with mutations of TP53 are common in therapy-related MDS and AML after therapy with alkylating agents: An M-FISH study. Genes Chromosomes Cancer 2005, 42, 358–371. [Google Scholar] [CrossRef]
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Kuendgen, A.; Nomdedeu, M.; Tuechler, H.; Garcia-Manero, G.; Komrokji, R.S.; Sekeres, M.A.; Della Porta, M.G.; Cazzola, M.; DeZern, A.E.; Roboz, G.J.; et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification—an approach to classification of patients with t-MDS. Leukemia 2021, 35, 835–849. [Google Scholar] [CrossRef]
- Awada, H.; Thapa, B.; Visconte, V. The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications. Cells 2020, 9, 2512. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Dragoljevic, D.; Westerterp, M.; Veiga, C.B.; Nagareddy, P.; Murphy, A.J. Disordered haematopoiesis and cardiovascular disease: A focus on myelopoiesis. Clin. Sci. 2018, 132, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R. Implications of molecular genetic diversity in myelodysplastic syndromes. Curr. Opin. Hematol. 2017, 24, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3699. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Makishima, H.; Kerr, C.M.; Przychodzen, B.P.; Aly, M.; Goyal, A.; Awada, H.; Asad, M.F.; Kuzmanovic, T.; Suzuki, H.; et al. Invariant patterns of clonal succession determine specific clinical features of myelodysplastic syndromes. Nat. Commun. 2019, 10, 5386. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awada, H.; Nagata, Y.; Goyal, A.; Asad, M.F.; Patel, B.; Hirsch, C.M.; Kuzmanovic, T.; Guan, Y.; Przychodzen, B.P.; Aly, M.; et al. Invariant phenotype and molecular association of biallelic TET2 mutant myeloid neoplasia. Blood Adv. 2019, 3, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, C.M.; Nazha, A.; Kneen, K.; Abazeed, M.E.; Meggendorfer, M.; Przychodzen, B.P.; Nadarajah, N.; Adema, V.; Nagata, Y.; Goyal, A.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia 2018, 32, 1751–1761. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Friesen, I.; Damm, F.; Yun, H.; Weissinger, E.M.; Krauter, J.; Wagner, K.; Chaturvedi, A.; Sharma, A.; Wichmann, M.; et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2499–2506. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdelwahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241.e228. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Tebaldi, T.; Rejeski, K.; Joshi, P.; Stefani, G.; Taylor, A.; Song, Y.; Vasic, R.; Maziarz, J.; Balasubramanian, K.; et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 2018, 32, 2659–2671. [Google Scholar] [CrossRef]
- Harada, Y.; Inoue, D.; Ding, Y.; Imagawa, J.; Doki, N.; Matsui, H.; Yahata, T.; Matsushita, H.; Ando, K.; Sashida, G.; et al. RUNX1/AML1 mutant collaborates with BMI1 overexpression in the development of human and murine myelodysplastic syndromes. Blood 2013, 121, 3434–3446. [Google Scholar] [CrossRef] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Lieschke, E.; Wang, Z.; Kelly, G.L.; Strasser, A. Discussion of some ‘knowns’ and some ‘unknowns’ about the tumour suppressor p53. J. Mol. Cell Biol. 2018, 11, 212–223. [Google Scholar] [CrossRef]
- Kon, A.; Shih, L.-Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef] [Green Version]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef] [Green Version]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018, 3, 46. [Google Scholar] [CrossRef]
- Hollink, I.; van den Ouweland, A.M.W.; Beverloo, H.B.; Arentsen-Peters, S.; Zwaan, C.M.; Wagner, A. Acute myeloid leukaemia in a case with Tatton-Brown-Rahman syndrome: The peculiar DNMT3A R882 mutation. J. Med. Genet. 2017, 54, 805–808. [Google Scholar] [CrossRef]
- Jacobs, R.H.; Cornbleet, M.A.; Vardiman, J.W.; Larson, R.A.; Le Beau, M.M.; Rowley, J.D. Prognostic implications of morphology and karyotype in primary myelodysplastic syndromes. Blood 1986, 67, 1765–1772. [Google Scholar] [CrossRef]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [Green Version]
- Sebaa, A.; Ades, L.; Baran-Marzack, F.; Mozziconacci, M.J.; Penther, D.; Dobbelstein, S.; Stamatoullas, A.; Récher, C.; Prebet, T.; Moulessehoul, S.; et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer 2012, 51, 1086–1092. [Google Scholar] [CrossRef]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes With del(5q) Predict Disease Progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef]
- Kere, J.; Ruutu, T.; de la Chapelle, A. Monosomy 7 in Granulocytes and Monocytes in Myelodysplastic Syndrome. N. Engl. J. Med. 1987, 316, 499–503. [Google Scholar] [CrossRef]
- Shiseki, M.; Ishii, M.; Okada, M.; Ohwashi, M.; Wang, Y.H.; Osanai, S.; Yoshinaga, K.; Mori, N.; Motoji, T.; Tanaka, J. Expression analysis of genes located within the common deleted region of del(20q) in patients with myelodysplastic syndromes. Leuk. Res. 2019, 84, 106175. [Google Scholar] [CrossRef]
- Caponetti, G.C.; Bagg, A. Mutations in myelodysplastic syndromes: Core abnormalities and CHIPping away at the edges. Int. J. Lab. Hematol. 2020, 42, 671–684. [Google Scholar] [CrossRef]
- Balasubramanian, S.K.; Aly, M.; Nagata, Y.; Bat, T.; Przychodzen, B.P.; Hirsch, C.M.; Adema, V.; Visconte, V.; Kuzmanovic, T.; Radivoyevitch, T.; et al. Distinct clinical and biological implications of various DNMT3A mutations in myeloid neoplasms. Leukemia 2018, 32, 550–553. [Google Scholar] [CrossRef]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and Maintenance of Genomic Methylation Patterns in Mouse Embryonic Stem Cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Guermouche, H.; Ravalet, N.; Gallay, N.; Deswarte, C.; Foucault, A.; Beaud, J.; Rault, E.; Saindoy, E.; Lachot, S.; Martignoles, J.-A.; et al. High prevalence of clonal hematopoiesis in the blood and bone marrow of healthy volunteers. Blood Adv. 2020, 4, 3550–3557. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Izzo, F.; Lee, S.C.; Poran, A.; Chaligne, R.; Gaiti, F.; Gross, B.; Murali, R.R.; Deochand, S.D.; Ang, C.; Jones, P.W.; et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020, 52, 378–387. [Google Scholar] [CrossRef]
- Dai, Y.J.; Wang, Y.Y.; Huang, J.Y.; Xia, L.; Shi, X.D.; Xu, J.; Lu, J.; Su, X.B.; Yang, Y.; Zhang, W.N.; et al. Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. USA 2017, 114, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kim, Y.R.; Yoo, N.J.; Lee, S.H. Mutational analysis of DNMT3A gene in acute leukemias and common solid cancers. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2013, 121, 85–94. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Yao, S.; Wang, Y.; Rolfe, A.; Selvaraj, A.; Darman, R.; Ke, J.; Warmuth, M.; Smith, P.G.; Larsen, N.A.; et al. The R882H DNMT3A hot spot mutation stabilizes the formation of large DNMT3A oligomers with low DNA methyltransferase activity. J. Biol. Chem. 2019, 294, 16966–16977. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wang, J.; Ren, Z.; Yin, J.; Wang, Y.; Cai, L.; Wang, G.G. A Model System for Studying the DNMT3A Hotspot Mutation (DNMT3AR882) Demonstrates a Causal Relationship between Its Dominant-Negative Effect and Leukemogenesis. Cancer Res. 2019, 79, 3583. [Google Scholar] [CrossRef]
- Emperle, M.; Dukatz, M.; Kunert, S.; Holzer, K.; Rajavelu, A.; Jurkowska, R.Z.; Jeltsch, A. The DNMT3A R882H mutation does not cause dominant negative effects in purified mixed DNMT3A/R882H complexes. Sci. Rep. 2018, 8, 13242. [Google Scholar] [CrossRef]
- Emperle, M.; Adam, S.; Kunert, S.; Dukatz, M.; Baude, A.; Plass, C.; Rathert, P.; Bashtrykov, P.; Jeltsch, A. Mutations of R882 change flanking sequence preferences of the DNA methyltransferase DNMT3A and cellular methylation patterns. Nucleic Acids Res. 2019, 47, 11355–11367. [Google Scholar] [CrossRef] [Green Version]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-mediated gene editing to assess the roles of TET2 and DNMT3A in clonal hematopoiesis and cardiovascular disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Ito, K.; Lee, J.; Chrysanthou, S.; Zhao, Y.; Josephs, K.; Sato, H.; Teruya-Feldstein, J.; Zheng, D.; Dawlaty, M.M.; Ito, K. Non-catalytic Roles of Tet2 Are Essential to Regulate Hematopoietic Stem and Progenitor Cell Homeostasis. Cell Rep. 2019, 28, 2480–2490. [Google Scholar] [CrossRef]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Asai, T.; Liu, Y.; Di Giandomenico, S.; Bae, N.; Ndiaye-Lobry, D.; Deblasio, A.; Menendez, S.; Antipin, Y.; Reva, B.; Wevrick, R.; et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood 2012, 120, 1601–1612. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gao, R.; Yao, C.; Kobayashi, M.; Liu, S.Z.; Yoder, M.C.; Broxmeyer, H.; Kapur, R.; Boswell, H.S.; Mayo, L.D.; et al. Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia 2018, 32, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Bondar, T.; Medzhitov, R. p53-Mediated Hematopoietic Stem and Progenitor Cell Competition. Cell Stem Cell 2010, 6, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; Porter, C.C.; Zaberezhnyy, V.; DeGregori, J. Irradiation Selects for p53-Deficient Hematopoietic Progenitors. PLoS Biol. 2010, 8, e1000324. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Rocquain, J.; Carbuccia, N.; Trouplin, V.; Raynaud, S.; Murati, A.; Nezri, M.; Tadrist, Z.; Olschwang, S.; Vey, N.; Birnbaum, D.; et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010, 10, 401. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 2010, 24, 1062–1065. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.-A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Jani, K.S.; Jain, S.U.; Ge, E.J.; Diehl, K.L.; Lundgren, S.M.; Müller, M.M.; Lewis, P.W.; Muir, T.W. Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc. Natl. Acad. Sci. USA 2019, 116, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Dewald, G.W.; Lasho, T.L.; Powell, H.L.; McClure, R.F.; Levine, R.L.; Gilliland, D.G.; Tefferi, A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood 2005, 106, 1207–1209. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Wang, Y.; Yura, Y.; Sano, M.; Oshima, K.; Yang, Y.; Katanasaka, Y.; Min, K.-D.; Matsuura, S.; Ravid, K.; et al. JAK2 (V617F) -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl. Sci. 2019, 4, 684–697. [Google Scholar] [CrossRef]
- Ko, M.; Rao, A. TET2: Epigenetic safeguard for HSC. Blood 2011, 118, 4501–4503. [Google Scholar] [CrossRef]
- Tung-Liang, L.; Yasunobu, N.; Hsiao-Wen, K.; Masashi, S.; Yusuke, O.; Chein-Fuang, H.; Der-Cherng, L.; Ming-Chung, K.; Chang-Liang, L.; En-Hui, L.; et al. Clonal leukemic evolution in myelodysplastic syndromes with TET2 and IDH1/2 mutations. Haematologica 2014, 99, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; He, X.; Zhu, Y.; Ding, Z.; Dong, H.; Feng, Y.; Du, J.; Wang, H.; Wu, X.; Zhang, L.; et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell Stem Cell 2018, 23, 355–369.e359. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e374. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.N.; Miller, C.A.; Jotte, M.R.M.; Bagegni, N.; Baty, J.D.; Schmidt, A.P.; Cashen, A.F.; Duncavage, E.J.; Helton, N.M.; Fiala, M.; et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 2018, 9, 455. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Takahashi, K.; Patel, K.; Bueso-Ramos, C.; Zhang, J.; Gumbs, C.; Jabbour, E.; Kadia, T.; Andreff, M.; Konopleva, M.; DiNardo, C.; et al. Clinical implications of TP53 mutations in myelodysplastic syndromes treated with hypomethylating agents. Oncotarget 2016, 7, 14172–14187. [Google Scholar] [CrossRef] [Green Version]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gäken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B.; et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Ebert, B.L. Molecular Pathophysiology of Myelodysplastic Syndromes. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 21–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Park, U.-H.; Yoon, S.K.; Park, T.; Kim, E.-J.; Um, S.-J. Additional sex comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor {gamma}. J. Biol. Chem. 2011, 286, 1354–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Bai, H.; Megyola, C.M.; Halene, S.; Krause, D.S.; Scadden, D.T.; Lu, J. Complex oncogene dependence in microRNA-125a-induced myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2012, 109, 16636–16641. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 leads to myelodysplastic syndrome–like disease in mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; Lafave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Group, U.S.S.; Crawley, C.; Craig, J.; Scott, M.A.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Ihle, J.N.; Kerr, I.M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995, 11, 69–74. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Martin, A.; Hasan, S.; Jacquot, S.; Birling, M.-C.; Vainchenker, W.; Villeval, J.-L. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood 2010, 116, 783–787. [Google Scholar] [CrossRef]
- Akada, H.; Yan, D.; Zou, H.; Fiering, S.; Hutchison, R.E.; Mohi, M.G. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera–like disease. Blood 2010, 115, 3589–3597. [Google Scholar] [CrossRef] [Green Version]
- Flynn, M.C.; Kraakman, M.J.; Tikellis, C.; Lee, M.K.S.; Hanssen, N.M.J.; Kammoun, H.L.; Pickering, R.J.; Dragoljevic, D.; Al-Sharea, A.; Barrett, T.J.; et al. Transient Intermittent Hyperglycemia Accelerates Atherosclerosis by Promoting Myelopoiesis. Circ. Res. 2020, 127, 877–892. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayyani, F.; Conley, A.P.; Strom, S.S.; Stevenson, W.; Cortes, J.E.; Borthakur, G.; Faderl, S.; O’Brien, S.; Pierce, S.; Kantarjian, H.; et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer 2010, 116, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Adrianzen Herrera, D.; Pradhan, K.; Snyder, R.; Karanth, S.; Janakiram, M.; Mantzaris, I.; Braunschweig, I.; Budhathoki, A.; Shah, U.A.; Verma, A.K.; et al. Myelodysplastic syndromes and the risk of cardiovascular disease in older adults: A SEER-medicare analysis. Leukemia 2020, 34, 1689–1693. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Murphy, A.J.; Tall, A.R. Disordered haematopoiesis and athero-thrombosis. Eur. Heart J. 2016, 37, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Sallman, D.A.; List, A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Diagnostic Criteria for MDS and Pre-Cursor Conditions (Defined by the WHO) [1,7] | |
|---|---|
| MDS | Persistent cytopenia in one or more peripheral-blood cell lineages and morphologic dysplasia (>10% dysplastic cells) in one or more bone marrow lineages on the basis of morphologic and cytogenetic abnormalities |
| MDS categories | MDS with: ● Single-lineage dysplasia ● Multilineage dysplasia ● Ring sideroblasts and single-lineage dysplasia or multilineage dysplasia ● Isolated del (5q) ● Excess blasts type 1 or type 2 ● Unclassifiable |
| CCUS | Unexplained cytopenia in one or more peripheral blood cell lineages; a somatic mutation at a variant allele frequency of at least 20% in one or more genes that are recurrently mutated in myeloid neoplasms and insufficient dysplasia (<10%) for an MDS diagnosis |
| CHIP | Normal peripheral blood cell counts with a somatic mutation at a variant allele frequency of at least 2% in a gene that is recurrently mutated in myeloid neoplasms |
| Summary of the Genetic Causes of MDS | |
|---|---|
| Somatic Mutations | |
| Epigenetic Regulators | TET2, DNMT3A, ASXL1, EZH2 genes |
| RNA Spliceosome | SF3B1, SRSF2, U2AF1 genes |
| DNA Transcription | RUNX1, TP53 genes |
| Signal Transduction Pathways | KRAS, NRAS, JAK2 genes |
| Cohesion complex | SMC3, SMC1A, RAD21, STAG2 genes |
| Germline Mutations | |
| CEBPA, DDX41, ETV6, GATA2, RUNX1 genes | |
| Inherited Disorders | |
| Fanconi Anemia | FANC genes |
| Shwachman-Diamond Syndrome | SBDS genes |
| Li-Fraumeni Syndrome | TP53 gene |
| Diamond-Blackfan Amemia | GATA1/RPS19 genes |
| Dyskeratosis congenita | Telomerase complex disorder |
| Chromosomal Abnormalities | |
| Chromosomal Deletion | del(5q), del(7q), del(20q), del(17q) |
| Mosaicism syndromes | Trisomy 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veiga, C.B.; Lawrence, E.M.; Murphy, A.J.; Herold, M.J.; Dragoljevic, D. Myelodysplasia Syndrome, Clonal Hematopoiesis and Cardiovascular Disease. Cancers 2021, 13, 1968. https://doi.org/10.3390/cancers13081968
Veiga CB, Lawrence EM, Murphy AJ, Herold MJ, Dragoljevic D. Myelodysplasia Syndrome, Clonal Hematopoiesis and Cardiovascular Disease. Cancers. 2021; 13(8):1968. https://doi.org/10.3390/cancers13081968
Chicago/Turabian StyleVeiga, Camilla Bertuzzo, Erin M. Lawrence, Andrew J. Murphy, Marco J. Herold, and Dragana Dragoljevic. 2021. "Myelodysplasia Syndrome, Clonal Hematopoiesis and Cardiovascular Disease" Cancers 13, no. 8: 1968. https://doi.org/10.3390/cancers13081968
APA StyleVeiga, C. B., Lawrence, E. M., Murphy, A. J., Herold, M. J., & Dragoljevic, D. (2021). Myelodysplasia Syndrome, Clonal Hematopoiesis and Cardiovascular Disease. Cancers, 13(8), 1968. https://doi.org/10.3390/cancers13081968