
A Phase 2 Clinical Trial of Trametinib and Low-Dose Dabrafenib in Patients with Advanced Pretreated NRASQ61R/K/L Mutant Melanoma (TraMel-WT)

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Design and Patient Population
2.2. Procedures and Study Treatment
2.3. Endpoints
2.4. ctDNA Analysis
2.5. Total Metabolic Tumor Volume Analysis
2.6. Statistical Analysis
3. Results
3.1. Baseline Characteristics
3.2. Treatment Disposition
3.3. Safety
3.4. Efficacy
3.5. Exploratory Endpoints
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [Green Version]
- Weeraratna, A.T. RAF around the edges—The paradox of BRAF inhibitors. N. Engl. J. Med. 2012, 366, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E.; et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. 2012, 30, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Saab, K.R.; Mooradian, M.J.; Wang, D.Y.; Chon, J.; Xia, C.Y.; Bs, A.B.; Bs, K.T.A.; Menzies, A.M.; Johnson, D.B.; Sullivan, R.J.; et al. Tolerance and efficacy of BRAF plus MEK inhibition in patients with melanoma who previously have received programmed cell death protein 1-based therapy. Cancer 2019, 125, 884–891. [Google Scholar] [CrossRef]
- Schreuer, M.; Jansen, Y.; Planken, S.; Chevolet, I.; Seremet, T.; Kruse, V.; Neyns, B. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF(V600)-mutant melanoma: An open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017, 18, 464–472. [Google Scholar] [CrossRef]
- Awada, G.; Özdemir, I.; Schwarze, J.; Daeninck, E.; Gondry, O.; Jansen, Y.; Seremet, T.; Keyaerts, M.; Everaert, H.; Neyns, B. Baseline total metabolic tumor volume assessed by 18FDG-PET/CT predicts outcome in advanced melanoma patients treated with pembrolizumab. Ann. Oncol. 2018, 29, x7. [Google Scholar] [CrossRef]
- Awada, G.; Serruys, D.; Schwarze, J.K.; Van De Voorde, L.; Duerinck, J.; Neyns, B. Durable Complete Response of a Recurrent Mesencephalic Glioblastoma Treated with Trametinib and Low-Dose Dabrafenib in a Patient with Neurofibromatosis Type 1. Case Rep. Oncol. 2020, 13, 1031–1036. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
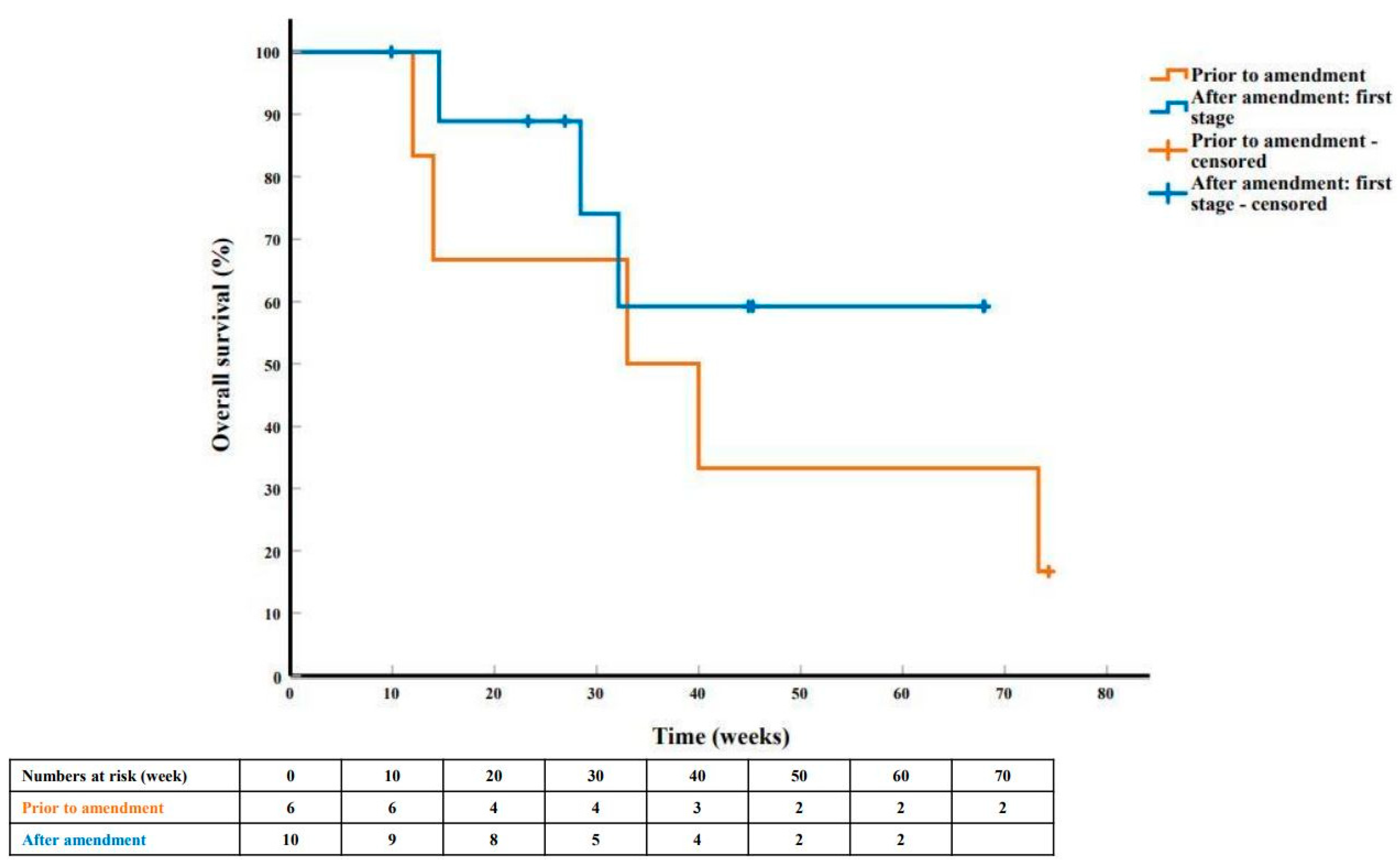{kind=link}
| Baseline Characteristics | Patients Enrolled Prior to Trial Amendment n = 6 | Patients Enrolled after Trial Amendment n = 10 |
|---|---|---|
| Sex (n (%)) | ||
| Male | 4 (66.7) | 4 (40.0) |
| Female | 2 (33.3) | 6 (60.0) |
| Age (median (range)) | 70 (44–77) | 58 (30–84) |
| ECOG PS (n (%)) | ||
| 0 | 5 (83.3) | 4 (40.0) |
| 1 | 1 (16.7) | 5 (50.0) |
| 2 | 0 | 1 (10.0) |
| Melanoma subtype (n (%)) | ||
| Superficial spreading | 2 (33.3) | 5 (50.0) |
| Nodular | 0 | 4 (40.0) |
| Lentigo maligna | 1 (16.7) | 0 |
| Spitzoid | 1 (16.7) | 0 |
| Cutaneous NOS | 1 (16.7) | 0 |
| Unknown primary lesion | 1 (16.7) | 1 (10.0) |
| AJCC stage (n (%)) | ||
| IIIC | 0 | 1 (10.0) |
| IV-M1b | 0 | 1 (10.0) |
| IV-M1c | 6 (100.0) | 5 (50.0) |
| IV-M1d | 0 | 3 (30.0) |
| Number of affected organs | ||
| Median (range) | 5 (2–7) | 5 (1–8) |
| Lactate dehydrogenase (n (%)) | ||
| Increased | 2 (33.3) | 4 (40.0) |
| Normal | 4 (66.7) | 6 (60.0) |
| NRAS mutation subtype (n (%)) | ||
| Q61R | 3 (50.0) | 6 (60.0) |
| Q61K | 3 (50.0) | 3 (30.0) |
| Q61L | 0 | 1 (10.0) |
| Prior lines of therapy | ||
| Median (range) | 2.5 (2–4) | 2.5 (1–5) |
| 1 (n (%)) | 0 | 3 (30.0) |
| 2 (n (%)) | 3 (50.0) | 5 (50.0) |
| 3 (n (%)) | 2 (33.3) | 0 |
| >3 (n (%)) | 1 (16.7) | 2 (20.0) |
| Prior PD-1 ICI (n (%)) | 6 (100.0) | 9 (90.0) |
| Prior CTLA-4 ICI (n (%)) | 5 (83.3) | 6 (60.0) |
| Prior PD-1 ICI/CTLA-4 ICI combination (n (%)) | 1 (16.7) | 3 (30.0) |
| NRASQ61R/K/L mutant ctDNA (n (%)) | ||
| Present | 2 (33.3) | 4 (40.0) |
| Absent | 4 (66.7) | 6 (60.0) |
| Total metabolic tumor volume mL (median (range)) | 123 (3–4392) | 44 (0–614) * |
| Adverse Events (n (%)) | Patients Enrolled Prior to Trial Amendment n = 6 | Patients Enrolled after Trial Amendment n = 10 | ||
|---|---|---|---|---|
| All-Grade | Grade 3–4 | All-Grade | Grade 3–4 | |
| All AE | 6 (100.0) | 3 (50.0) | 10 (100.0) | 4 (40.0) |
| Acneiform rash | 6 (100.0) * | 0 | 3 (30.0) ° | 0 |
| Acneiform rash leading to temporary treatment interruption | 6 (100.0) | 0 | 0 | 0 |
| Acneiform rash leading to add-on of low-dose dabrafenib | 6 (100.0) | 0 | NA | NA |
| Fatigue | 4 (66.7) | 1 (16.7) | 4 (40.0) | 0 |
| Creatine phosphokinase increase | 4 (66.7) | 0 | 6 (60.0) | 0 |
| Lipase increased | 3 (50.0) | 0 | 3 (30.0) | 0 |
| Anemia | 3 (50.0) | 0 | 2 (20.0) | 0 |
| Fever | 3 (50.0) | 0 | 1 (10.0) | 1 (10.0) |
| Arterial hypertension | 2 (33.3) | 0 | 3 (30.0) | 0 |
| Aspartate aminotransferase increase | 2 (33.3) | 0 | 3 (30.0) | 1 (10.0) |
| Central serous retinopathy | 2 (33.3) | 0 | 1 (10.0) | 0 |
| Diarrhea | 2 (33.3) | 0 | 4 (40.0) | 0 |
| Lymphocyte count decreased | 2 (33.3) | 0 | 1 (10.0) | 0 |
| Nausea | 2 (33.3) | 0 | 3 (30.0) | 0 |
| Hyperkalemia | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Hyponatremia | 1 (16.7) | 1 (16.7) | 3 (30.0) | 2 (20.0) |
| Idiopathic thrombocytopenic purpura | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Lung infection | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Syncope | 1 (16.7) | 1 (16.7) | 1 (10.0) | 1 (10.0) |
| Abdominal pain | 1 (16.7) | 0 | 1 (10.0) | 0 |
| Alanine aminotransferase increase | 1 (16.7) | 0 | 3 (30.0) | 2 (20.0) |
| Anorexia | 1 (16.7) | 0 | 2 (20.0) | 0 |
| Atrial fibrillation | 1 (16.7) | 0 | 0 | 0 |
| Blot bleed retina | 1 (16.7) | 0 | 0 | 0 |
| Chills | 1 (16.7) | 0 | 2 (20.0) | 0 |
| Cough | 1 (16.7) | 0 | 0 | 0 |
| Edema lower limbs | 1 (16.7) | 0 | 1 (10.0) | 0 |
| Eosinophilia | 1 (16.7) | 0 | 0 | 0 |
| Ejection fraction decreased | 1 (16.7) | 0 | 1 (10.0) | 1 (10.0) |
| Eosinophil count increased | 1 (16.7) | 0 | 0 | 0 |
| Heart failure | 1 (16.7) | 0 | 0 | 0 |
| Hypoalbuminemia | 1 (16.7) | 0 | 0 | 0 |
| Ileus | 1 (16.7) | 0 | 0 | 0 |
| Inflammatory syndrome | 1 (16.7) | 0 | 0 | 0 |
| Maculopapular rash | 1 (16.7) | 0 | 1 (10.0) | 0 |
| Malaise | 1 (16.7) | 0 | ||
| Neutrophil count decreased | 1 (16.7) | 0 | 3 (30.0) | 0 |
| Occlusion retinal arteriolus | 1 (16.7) | 0 | 0 | 0 |
| Operculum retinae | 1 (16.7) | 0 | 0 | 0 |
| Paronychia | 1 (16.7) | 0 | 0 | 0 |
| Platelet count decreased | 1 (16.7) | 0 | 2 (20.0) | 0 |
| Radiation pneumonitis | 1 (16.7) | 0 | 0 | 0 |
| White blood cell count decreased | 1 (16.7) | 0 | 2 (20.0) | 1 (10.0) |
| Vomiting | 0 | 0 | 3 (30.0) | 0 |
| Acute kidney injury | 0 | 0 | 2 (20.0) | 0 |
| Muscle cramps | 0 | 0 | 2 (20.0) | 0 |
| Gamma glutamyl transferase increased | 0 | 0 | 1 (10.0) | 1 (10.0) |
| Pneumonitis | 0 | 0 | 1 (10.0) | 1 (10.0) |
| Pulmonary embolism | 0 | 0 | 1 (10.0) | 1 (10.0) |
| Alkaline phosphatase increased | 0 | 0 | 1 (10.0) | 0 |
| Arthralgia | 0 | 0 | 1 (10.0) | 0 |
| Bronchopulmonary hemorrhage | 0 | 0 | 1 (10.0) | 0 |
| Dry mouth | 0 | 0 | 1 (10.0) | 0 |
| Dyspepsia | 0 | 0 | 1 (10.0) | 0 |
| Headache | 0 | 0 | 1 (10.0) | 0 |
| Hypokalemia | 0 | 0 | 1 (10.0) | 0 |
| Neuropathic pain | 0 | 0 | 1 (10.0) | 0 |
| Posterior vitreous detachment | 0 | 0 | 1 (10.0) | 0 |
| Retinal pigment epithelial detachment | 0 | 0 | 1 (10.0) | 0 |
| Retinoschisis | 0 | 0 | 1 (10.0) | 0 |
| Squamous rash | 0 | 0 | 1 (10.0) | 0 |
| Skin infection | 0 | 0 | 1 (10.0) | 0 |
| Urinary tract infection | 0 | 0 | 1 (10.0) | 0 |
| Serious AE | 3 (50.0) | 2 (33.3) | 5 (50.0) | 4 (40.0) |
| AE leading to dose reduction | 3 (50.0) | 0 | 5 (50.0) | 4 (40.0) |
| AE leading to temporary treatment interruption | 6 (100.0) | 2 (33.3) | 6 (60.0) | 4 (40.0) |
| AE leading to permanent treatment interruption | 0 | 0 | 0 | 0 |
| Best Objective Response (n (%)) | Patients Enrolled Prior to Trial Amendment n = 6 | Patients Enrolled after Trial Amendment n = 10 |
|---|---|---|
| Confirmed objective response | 1 (16.7) * | 0 |
| Complete response | 0 | 0 |
| Partial response | 1 (16.7) * | 0 |
| Stable disease | 3 (50.0) ° | 4 (40.0) |
| Progressive disease | 2 (33.3) | 6 (60.0) |
| Objective response rate | 1 (6.3) | |
| Disease control rate | 8 (50.0) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awada, G.; Schwarze, J.K.; Tijtgat, J.; Fasolino, G.; Everaert, H.; Neyns, B. A Phase 2 Clinical Trial of Trametinib and Low-Dose Dabrafenib in Patients with Advanced Pretreated NRASQ61R/K/L Mutant Melanoma (TraMel-WT). Cancers 2021, 13, 2010. https://doi.org/10.3390/cancers13092010
Awada G, Schwarze JK, Tijtgat J, Fasolino G, Everaert H, Neyns B. A Phase 2 Clinical Trial of Trametinib and Low-Dose Dabrafenib in Patients with Advanced Pretreated NRASQ61R/K/L Mutant Melanoma (TraMel-WT). Cancers. 2021; 13(9):2010. https://doi.org/10.3390/cancers13092010
Chicago/Turabian StyleAwada, Gil, Julia Katharina Schwarze, Jens Tijtgat, Giuseppe Fasolino, Hendrik Everaert, and Bart Neyns. 2021. "A Phase 2 Clinical Trial of Trametinib and Low-Dose Dabrafenib in Patients with Advanced Pretreated NRASQ61R/K/L Mutant Melanoma (TraMel-WT)" Cancers 13, no. 9: 2010. https://doi.org/10.3390/cancers13092010
APA StyleAwada, G., Schwarze, J. K., Tijtgat, J., Fasolino, G., Everaert, H., & Neyns, B. (2021). A Phase 2 Clinical Trial of Trametinib and Low-Dose Dabrafenib in Patients with Advanced Pretreated NRASQ61R/K/L Mutant Melanoma (TraMel-WT). Cancers, 13(9), 2010. https://doi.org/10.3390/cancers13092010