
Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors

 , , , and
, , , and 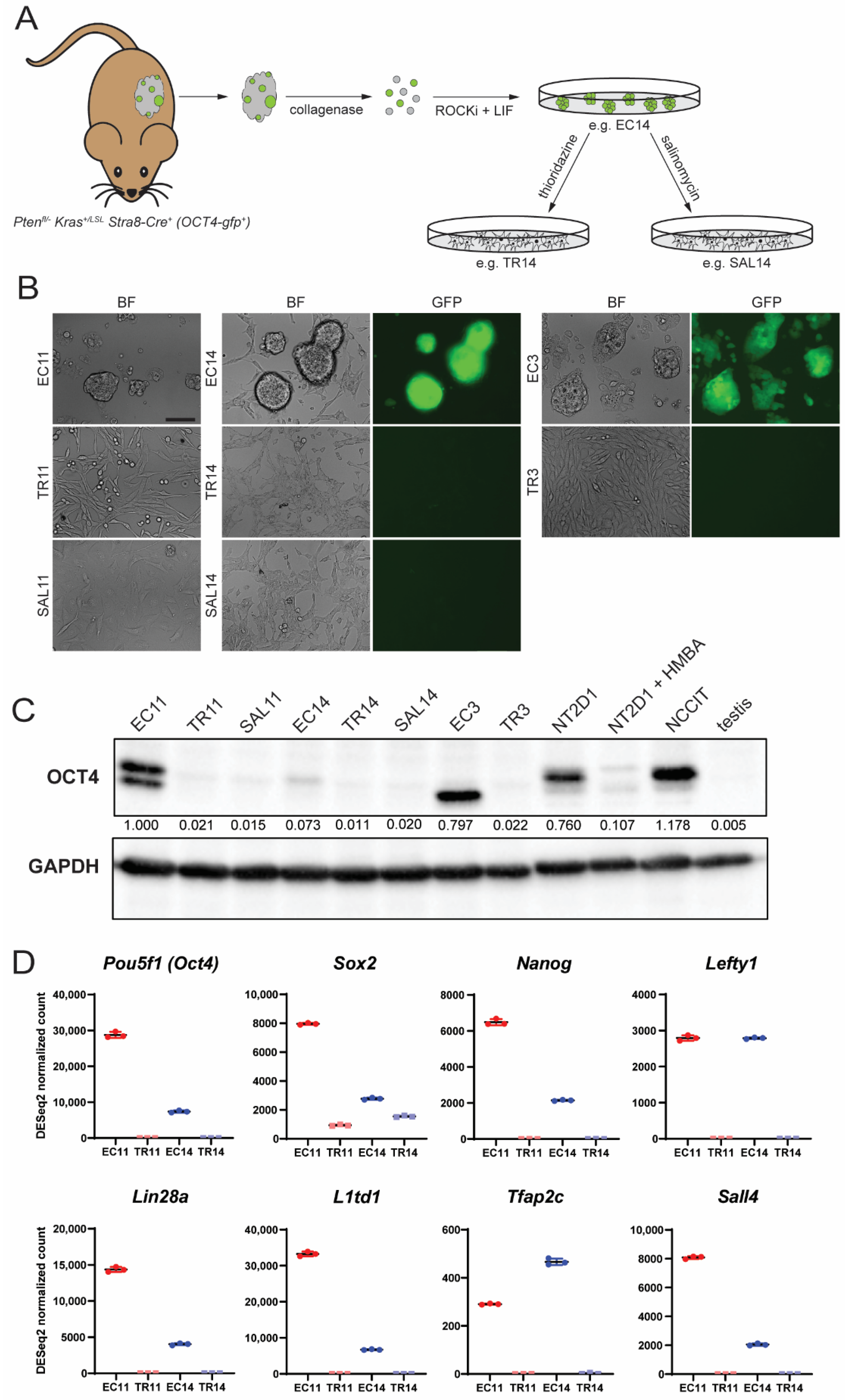{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Strains and Husbandry
2.2. Genotyping
2.3. Primary EC Cell Culture and Differentiation
2.4. Culture of Human EC Cell Lines
2.5. Derivation of Transformed iPS Cells
2.6. Western Blot Analysis
2.7. Quantitative PCR
2.8. RNA Sequencing
2.9. Gene Set Enrichment Analysis
2.10. Gene Ontology Analysis
2.11. Population Doubling Assay
2.12. Soft Agar Assay
2.13. Immunohistochemistry
2.14. Subcutaneous Cell Transplantations
2.15. Thioridazine Treatment of Tumor-Bearing Mice
2.16. Fertility Study
2.17. Statistical Analysis
3. Results
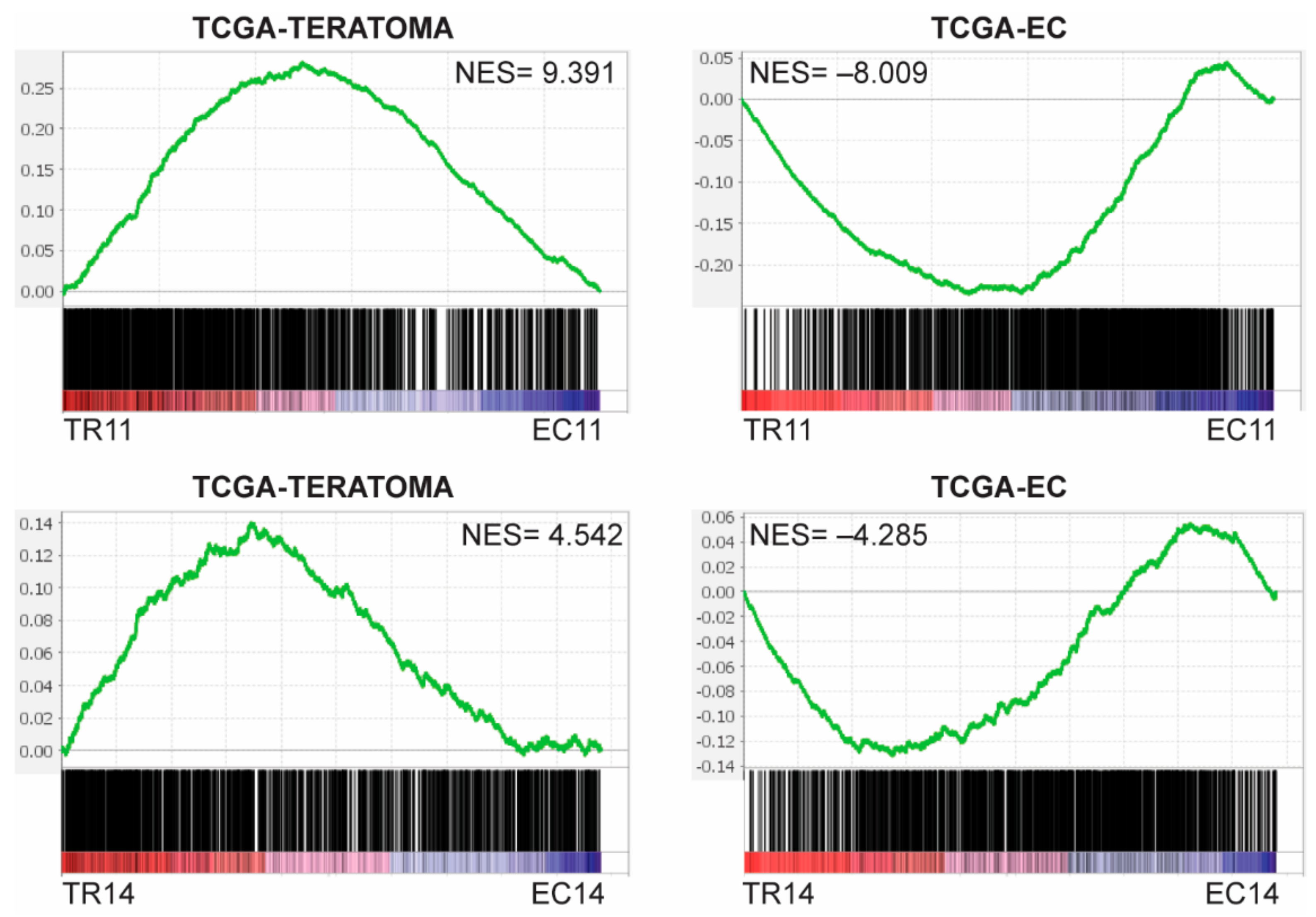
3.1. Thioridazine and Salinomycin Induce Differentiation of Murine EC Cells
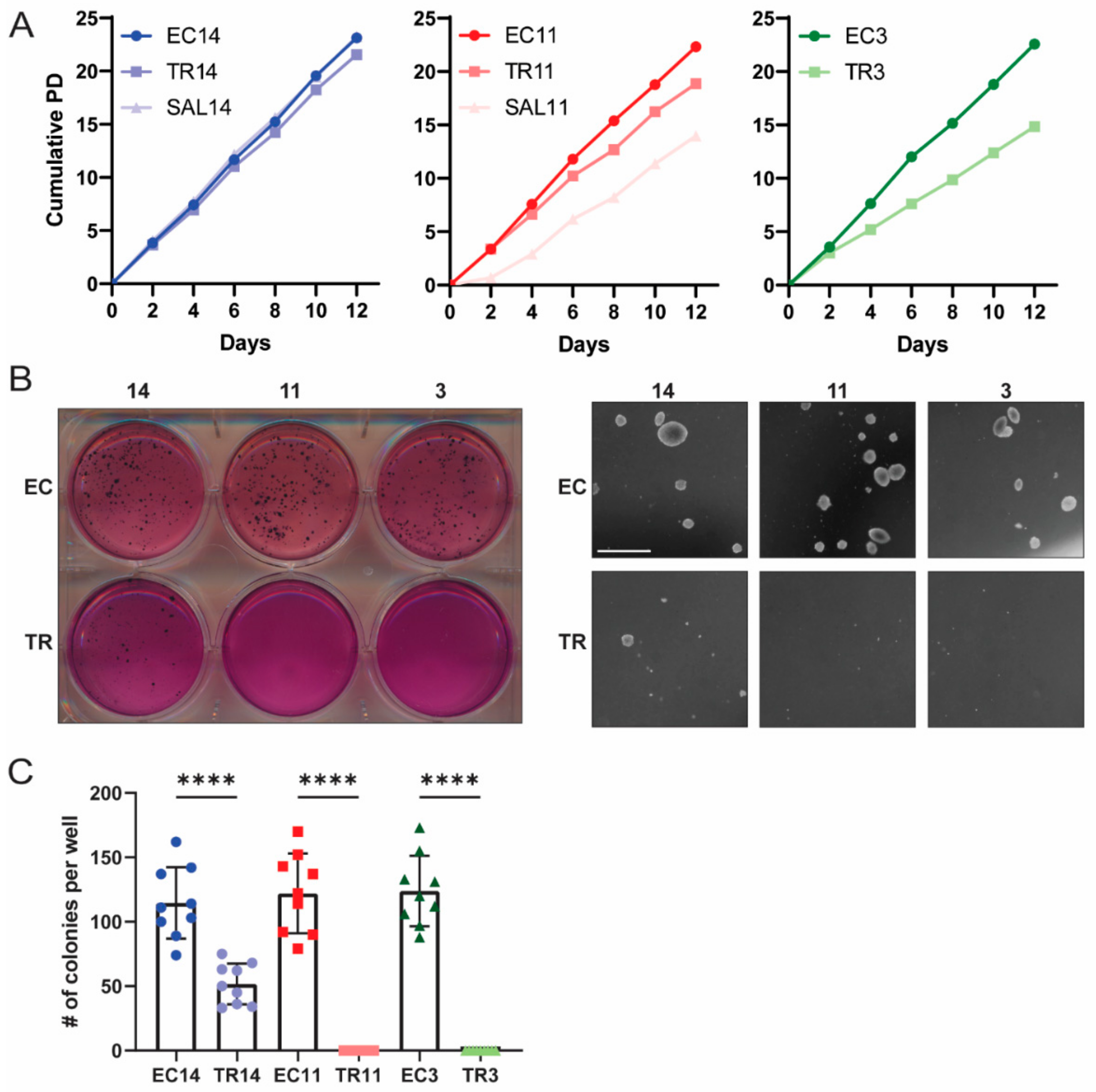
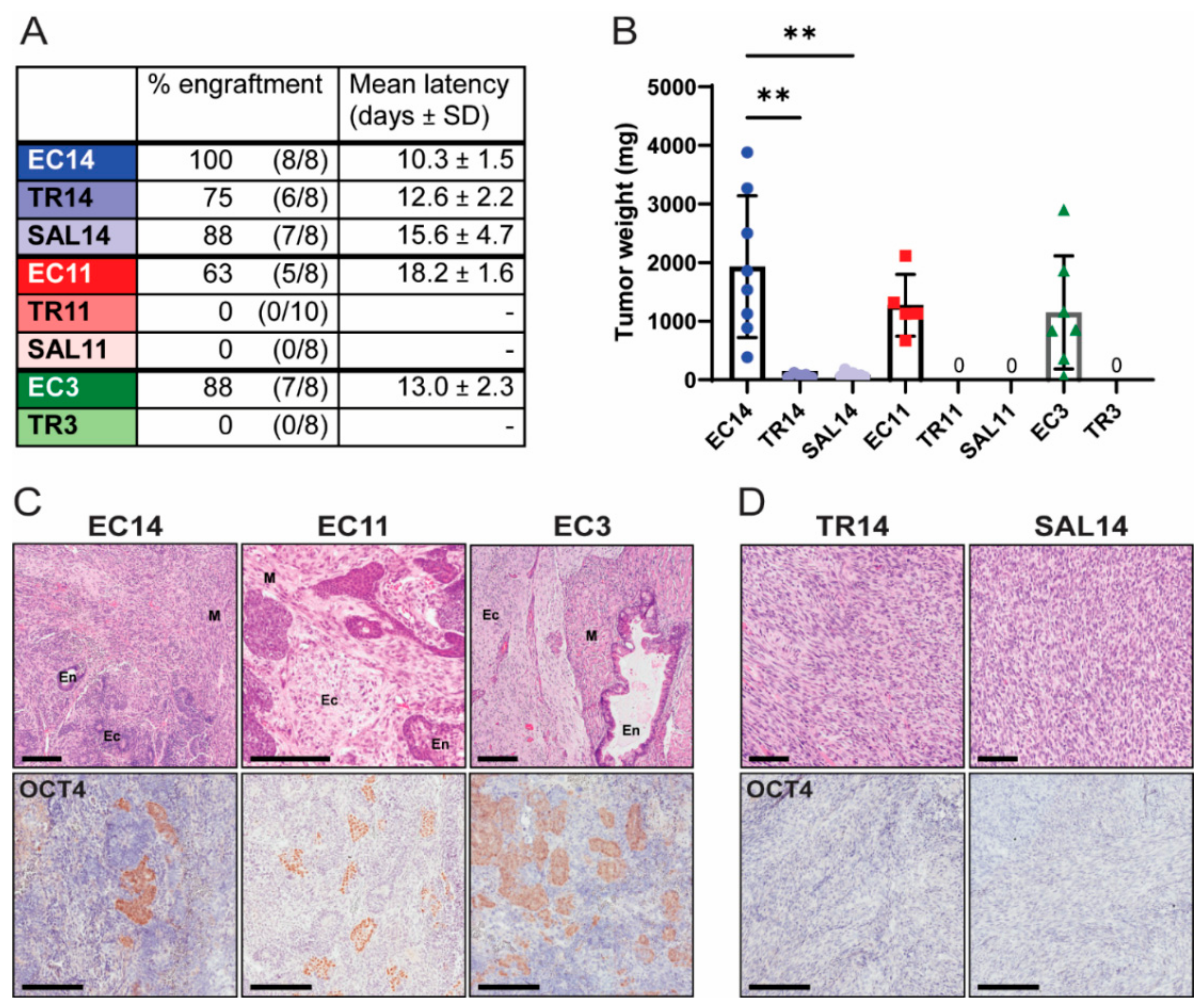
3.2. Thioridazine- and Salinomycin-Differentiated Cells Have a Reduced Tumorigenic Potential
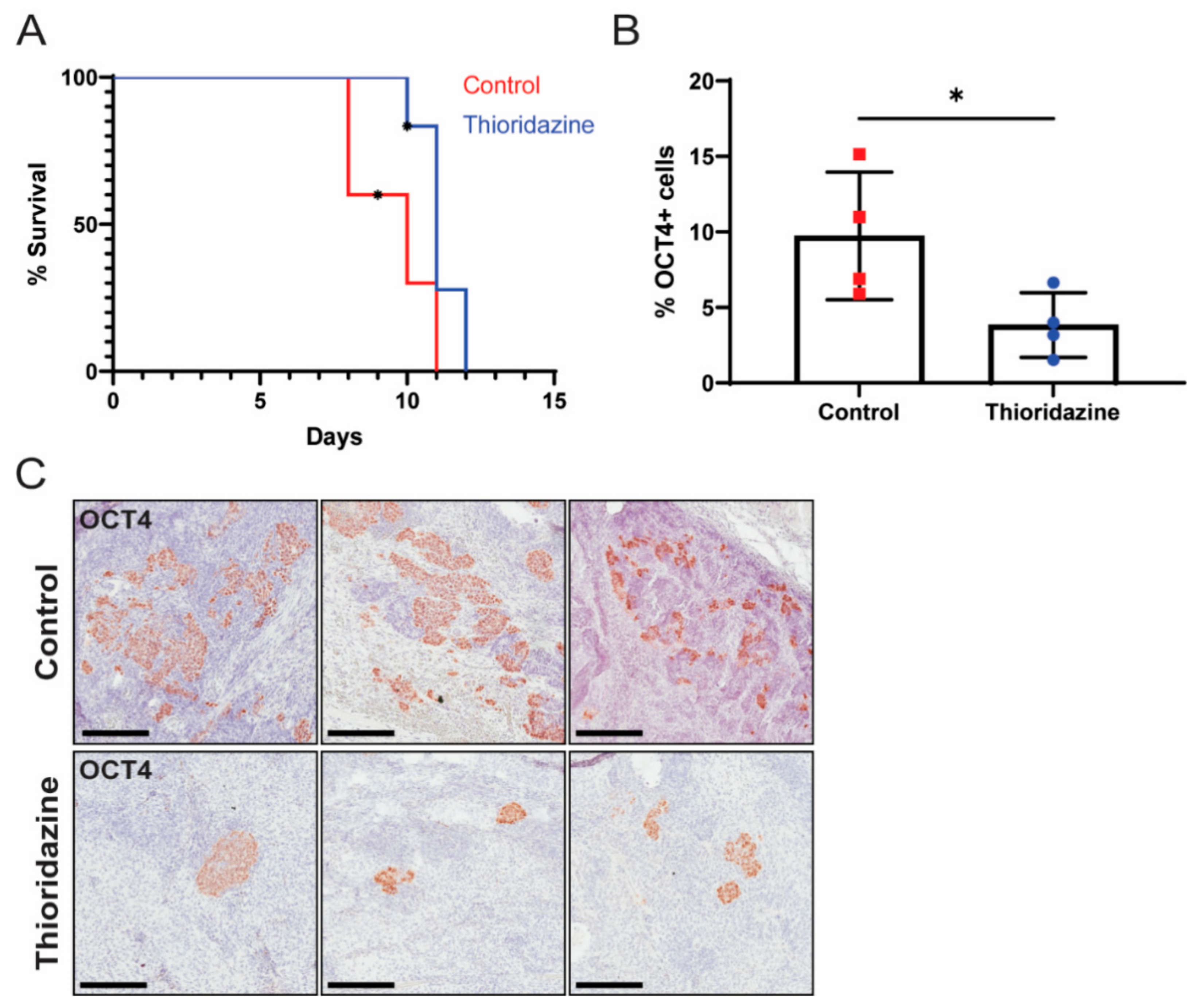
3.3. Thioridazine Treatment of Mice Bearing Transformed iPS Cell Allografts Reduces the Number of OCT4-Expressing Cells and Extends Survival
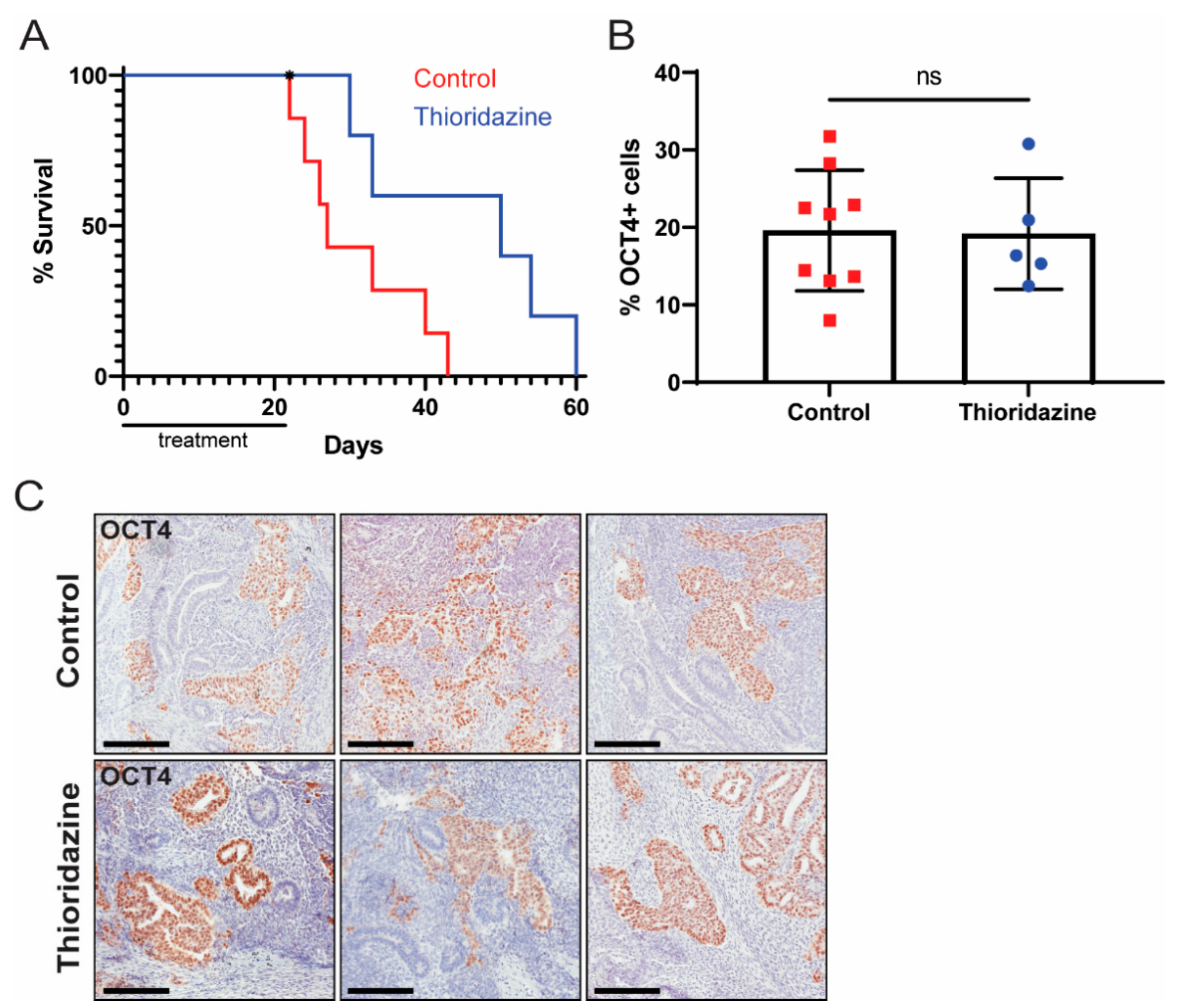
3.4. Thioridazine Treatment of Mice Bearing Human EC Cell Xenografts Extends Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winter, C.; Albers, P. Testicular germ cell tumors: Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2011, 7, 43–53. [Google Scholar] [CrossRef]
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L.H.J. Testicular cancer. Nat. Rev. Dis. Primers 2018, 4, 29. [Google Scholar] [CrossRef]
- Schmidtova, S.; Kalavska, K.; Kucerova, L. Molecular Mechanisms of Cisplatin Chemoresistance and Its Circumventing in Testicular Germ Cell Tumors. Curr. Oncol. Rep. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Lotan, T.L. The Lower Urinary Tract and Male Genital System. In Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Kumar, V., Abbas, A.K., Aster, J.C., Eds.; Elsevier/Saunders: Philadelphia, PA, USA, 2015; Chapter 21; pp. 959–990. [Google Scholar]
- Ulbright, T.M. Germ cell tumors of the gonads: A selective review emphasizing problems in differential diagnosis, newly appreciated, and controversial issues. Mod. Pathol. 2005, 18, S61–S79. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, G.; Gundgaard, M.G.; Mortensen, M.S.; Agerbæk, M.; Holm, N.V.; Rørth, M.; Von Der Maase, H.; Christensen, I.J.; Lauritsen, J. Surveillance for Stage I Nonseminoma Testicular Cancer: Outcomes and Long-Term Follow-Up in a Population-Based Cohort. J. Clin. Oncol. 2014, 32, 3817–3823. [Google Scholar] [CrossRef]
- Kier, M.G.; Lauritsen, J.; Mortensen, M.S.; Bandak, M.; Andersen, K.K.; Hansen, M.K.; Agerbaek, M.; Holm, N.V.; Dalton, S.O.; Johansen, C.; et al. Prognostic Factors and Treatment Results After Bleomycin, Etoposide, and Cisplatin in Germ Cell Cancer: A Population-based Study. Eur. Urol. 2017, 71, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Lauritsen, J.; Gundgaard, M.G.; Agerbæk, M.; Holm, N.V.; Christensen, I.J.; von der Maase, H.; Daugaard, G. A Nationwide Cohort Study of Stage I Seminoma Patients Followed on a Surveillance Program. Eur. Urol. 2014, 66, 1172–1178. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Meyts, E.R.-D.; McGlynn, K.A.; Okamoto, K.; Jewett, M.A.S.; Bokemeyer, C. Testicular germ cell tumours. Lancet 2016, 387, 1762–1774. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Dearnaley, D.P.; Huddart, R.A.; Horwich, A. Managing testicular cancer. BMJ 2001, 322, 1583. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Beard, C.; Allan, J.M.; Dahl, A.A.; Feldman, D.R.; Oldenburg, J.; Daugaard, G.; Kelly, J.L.; Dolan, M.E.; Hannigan, R.; et al. Testicular cancer survivorship: Research strategies and recommendations. J. Natl. Cancer Inst. 2010, 102, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Fossa, S.D.; Williams, A.; Travis, L.B. Long-term Morbidity of Testicular Cancer Treatment. Urol. Clin. N. Am. 2015, 42, 393–408. [Google Scholar] [CrossRef]
- Schepisi, G.; De Padova, S.; De Lisi, D.; Casadei, C.; Meggiolaro, E.; Ruffilli, F.; Rosti, G.; Lolli, C.; Ravaglia, G.; Conteduca, V.; et al. Psychosocial Issues in Long-Term Survivors of Testicular Cancer. Front. Endocrinol. 2019, 10, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, V.; Dinh, P.C.; Fung, C.; Monahan, P.O.; Althouse, S.K.; Norton, K.; Cary, C.; Einhorn, L.; Fossa, S.D.; Adra, N.; et al. Adverse Health Outcomes among US Testicular Cancer Survivors after Cisplatin-Based Chemotherapy vs Surgical Management. JNCI Cancer Spectr. 2020, 4, pkz079. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol./Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Sesterhenn, I.A.; Davis, J.C., Jr. Pathology of Germ Cell Tumors of the Testis. Cancer Control 2004, 11, 374–387. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-Induced Depletion of OCT4-Positive Cancer Stem Cells in a Mouse Model of Malignant Testicular Cancer. Cell Rep. 2017, 21, 1896–1909. [Google Scholar] [CrossRef] [Green Version]
- Batool, A.; Karimi, N.; Wu, X.-N.; Chen, S.-R.; Liu, Y.-X. Testicular germ cell tumor: A comprehensive review. Cell. Mol. Life Sci. 2019, 76, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Kevin, D.A., II; Meujo, D.A.; Hamann, M.T. Polyether ionophores: Broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites. Expert Opin. Drug Discov. 2009, 4, 109–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachlos, E.; Risueño, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.-H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of Drugs Including a Dopamine Receptor Antagonist that Selectively Target Cancer Stem Cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driver, J.A.; Logroscino, G.; Buring, J.E.; Gaziano, J.M.; Kurth, T. A Prospective Cohort Study of Cancer Incidence Following the Diagnosis of Parkinson’s Disease. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Dalton, S.O.; Mellemkjær, L.; Thomassen, L.; Mortensen, P.B.; Johansen, C. Risk for cancer in a cohort of patients hospitalized for schizophrenia in Denmark, 1969–1993. Schizophr. Res. 2005, 75, 315–324. [Google Scholar] [CrossRef]
- Lyndaker, A.M.; Pierpont, T.M.; Loehr, A.R.; Weiss, R.S. A Genetically Engineered Mouse Model of Malignant Testicular Germ Cell Tumors. Methods Mol. Biol. 2021, 2195, 147–165. [Google Scholar] [CrossRef]
- National Research Council. Guide for the Care and Use of Laboratory Animals: Eighth Edition; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Schnabel, L.V.; Abratte, C.M.; Schimenti, J.C.; Southard, T.L.; Fortier, L.A. Genetic background affects induced pluripotent stem cell generation. Stem. Cell Res. Ther. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Weiss, R.S. Increased common fragile site expression, cell proliferation defects, and apoptosis following conditional inactivation of mouse Hus1 in primary cultured cells. Mol. Biol. Cell 2007, 18, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Gueguen, L.; Coppee, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Andrews, P.W.; Gonczol, E.; Plotkin, S.A.; Dignazio, M.; Oosterhuis, J.W. Differentiation of TERA-2 human embryonal carcinoma cells into neurons and HCMV permissive cells: Induction by agents other than retinoic acid. Differentiation 1986, 31, 119–126. [Google Scholar] [CrossRef]
- Mehravar, M.; Ghaemimanesh, F.; Poursani, E.M. An Overview on the Complexity of OCT4: At the Level of DNA, RNA and Protein. Stem Cell Rev. Rep. 2021. [Google Scholar] [CrossRef]
- Kleinsmith, L.J.; Pierce, G.B. Multipotentiality of Single Embryonal Carcinoma Cells. Cancer Res. 1964, 24, 1544–1551. [Google Scholar] [PubMed]
- Yong, M.; Yu, T.; Tian, S.; Liu, S.; Xu, J.; Hu, J.; Hu, L. DR2 blocker thioridazine: A promising drug for ovarian cancer therapy. Oncol. Lett. 2017, 14, 8171–8177. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Damjanov, I.; Simon, D.; Banting, G.S.; Carlin, C.; Dracopoli, N.C.; Fogh, J. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Investig. 1984, 50, 147–162. [Google Scholar]
- Varga, B.; Csonka, Á.; Csonka, A.; MolnÁR, J.; Amaral, L.; Spengler, G. Possible Biological and Clinical Applications of Phenothiazines. Anticancer Res. 2017, 37, 5983–5993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohle, G.R. Male infertility in cancer patients: Review of the literature. Int. J. Urol. 2010, 17, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Cabral, F.C.; Krajewski, K.M.; Rosenthal, M.H.; Hirsch, M.S.; Howard, S.A. Teratoma with malignant transformation: Report of three cases and review of the literature. Clin. Imaging 2014, 38, 589–593. [Google Scholar] [CrossRef]
- Strickland, S.; Mahdavi, V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978, 15, 393–403. [Google Scholar] [CrossRef]
- Castaigne, S.; Chomienne, C.; Daniel, M.T.; Ballerini, P.; Berger, R.; Fenaux, P.; Degos, L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 1990, 76, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef]
- Mueller, T.; Mueller, L.P.; Luetzkendorf, J.; Voigt, W.; Simon, H.; Schmoll, H.J. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumor Biol. 2006, 27, 71–83. [Google Scholar] [CrossRef]
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem. Cell Biol. 2010, 134, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Dai, L.; Yu, T. Thioridazine Sensitizes Cisplatin against Chemoresistant Human Lung and Ovary Cancer Cells. DNA Cell Biol. 2019, 38, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Hasan-Olive, M.M.; Zhu, H.; Denisova, O.; Grudic, A.; Latif, M.A.; Saed, H.; Varughese, J.K.; Rosland, G.V.; Yang, N.; et al. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int. J. Cancer 2019, 144, 1735–1745. [Google Scholar] [CrossRef]
- Seo, S.U.; Cho, H.K.; Min, K.J.; Woo, S.M.; Kim, S.; Park, J.W.; Kim, S.H.; Choi, Y.H.; Keum, Y.S.; Hyun, J.W.; et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death Dis. 2017, 8, e2599. [Google Scholar] [CrossRef]
- Tegowski, M.; Fan, C.; Baldwin, A.S. Thioridazine inhibits self-renewal in breast cancer cells via DRD2-dependent STAT3 inhibition, but induces a G1 arrest independent of DRD2. J. Biol. Chem. 2018, 293, 15977–15990. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.W.; Ko, H.J.; Chou, C.H.; Cheng, T.S.; Cheng, H.W.; Liang, Y.H.; Lai, Y.L.; Lin, C.Y.; Wang, C.; Loh, J.K.; et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Dong, S.M.; Kim, B.R.; Park, M.S.; Trink, B.; Byun, H.J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Aslostovar, L.; Boyd, A.L.; Almakadi, M.; Collins, T.J.; Leong, D.P.; Tirona, R.G.; Kim, R.B.; Julian, J.A.; Xenocostas, A.; Leber, B.; et al. A phase 1 trial evaluating thioridazine in combination with cytarabine in patients with acute myeloid leukemia. Blood Adv. 2018, 2, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Aslostovar, L.; Boyd, A.L.; Benoit, Y.D.; Di Lu, J.; Rodriguez, J.L.G.; Nakanishi, M.; Porras, D.P.; Reid, J.C.; Mitchell, R.R.; Leber, B.; et al. Abnormal dopamine receptor signaling allows selective therapeutic targeting of neoplastic progenitors in AML patients. Cell Rep. Med. 2021, 2, 100202. [Google Scholar] [CrossRef]
- Lee, S.I.; Roney, M.S.; Park, J.H.; Baek, J.Y.; Park, J.; Kim, S.K.; Park, S.K. Dopamine receptor antagonists induce differentiation of PC-3 human prostate cancer cell-derived cancer stem cell-like cells. Prostate 2019, 79, 720–731. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loehr, A.R.; Pierpont, T.M.; Gelsleichter, E.; Galang, A.M.D.; Fernandez, I.R.; Moore, E.S.; Guo, M.Z.; Miller, A.D.; Weiss, R.S. Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors. Cancers 2021, 13, 2045. https://doi.org/10.3390/cancers13092045
Loehr AR, Pierpont TM, Gelsleichter E, Galang AMD, Fernandez IR, Moore ES, Guo MZ, Miller AD, Weiss RS. Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors. Cancers. 2021; 13(9):2045. https://doi.org/10.3390/cancers13092045
Chicago/Turabian StyleLoehr, Amanda R., Timothy M. Pierpont, Eric Gelsleichter, Anabella Maria D. Galang, Irma R. Fernandez, Elizabeth S. Moore, Matthew Z. Guo, Andrew D. Miller, and Robert S. Weiss. 2021. "Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors" Cancers 13, no. 9: 2045. https://doi.org/10.3390/cancers13092045
APA StyleLoehr, A. R., Pierpont, T. M., Gelsleichter, E., Galang, A. M. D., Fernandez, I. R., Moore, E. S., Guo, M. Z., Miller, A. D., & Weiss, R. S. (2021). Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors. Cancers, 13(9), 2045. https://doi.org/10.3390/cancers13092045