
Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression

 ,
,  and
and
Abstract
:Simple Summary
Abstract
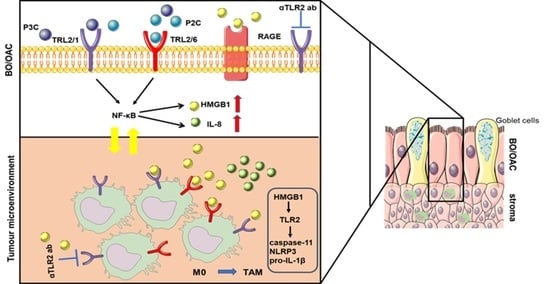
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Stimulation
2.2. Preparation of BMDMs
2.3. Murine Organoid Culture
2.4. Conditioned Media
2.5. QUANTI-BlueTM Assay
2.6. Detection of Supernatant Proteins
2.7. Isolation of Cytoplasmic Proteins
2.8. Immunoblotting
2.9. Cytokine Measurements
2.10. Quantitative Real-Time PCR
2.11. Statistical Analysis
3. Results
3.1. Cell Lines Derived from Barrett’s Dysplasia and Early Oesophageal Adenocarcinoma Are Responsive to TLR2
3.2. Oesophageal Cell Responses to TLR2 Stimulation Are Inhibited by a TLR2 Neutralising Antibody
3.3. TLR2 Inhibition Limits Chemokine Secretion from Oesophageal Organoids
3.4. Secretion of TLR2 Agonists by Oesophageal Cell Lines
3.5. Tumour Cells Release HMGB1 Following TLR2 Stimulation
3.6. TLR2-Stimulated EAC Cells Activate Macrophages through HMGB1.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maxwell Parkin, D.; Bray, F.; Ferlay, J.; Pisani, P. Estimating the world cancer burden: Globocan 2000. Int. J. Cancer 2001, 94, 153–156. [Google Scholar] [CrossRef]
- Thrift, A.P.; Whiteman, D.C. The incidence of esophageal adenocarcinoma continues to rise: Analysis of period and birth cohort effects on recent trends. Ann. Oncol. 2012, 23, 3155–3162. [Google Scholar] [CrossRef]
- Pera, M.; Manterola, C.; Vidal, O.; Grande, L. Epidemiology of esophageal adenocarcinoma. J. Surg. Oncol. 2005, 92, 151–159. [Google Scholar] [CrossRef]
- Yang, L.; Francois, F.; Pei, Z. Molecular Pathways: Pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett’s esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated with Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzka, D.A.; Falk, G.W. Management of Low-Grade Dysplasia in Barrett’s Esophagus: Incremental Progress Continues. Gastroenterology 2017, 152, 928–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Steffensen, K.D.; Mor, G. Cancers take their Toll—The function and regulation of Toll-like receptors in cancer cells. Oncogene 2008, 27, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Irvine, K.L.; Hopkins, L.J.; Gangloff, M.; Bryant, C.E. The molecular basis for recognition of bacterial ligands at equine TLR2, TLR1 and TLR6. Vet. Res. 2013, 44, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting Edge: Cardiac Myosin Activates Innate Immune Responses through Toll-like Receptors. J. Immunol. 2010, 183, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan Fragments Act as an Endogenous Danger Signal by Engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Vabulas, R.M.; Ahmad-Nejad, P.; Da Costa, C.; Miethke, T.; Kirschning, C.J.; Häcker, H.; Wagner, H. Endocytosed HSP60s Use Toll-like Receptor 2 (TLR2) and TLR4 to Activate the Toll/Interleukin-1 Receptor Signaling Pathway in Innate Immune Cells. J. Biol. Chem. 2001, 276, 31332–31339. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Huhta, H.; Helminen, O.; Lehenkari, P.P.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Toll-like receptors 1, 2, 4 and 6 in esophageal epithelium, Barrett’s esophagus, dysplasia and adenocarcinoma. Oncotarget 2016, 7, 23658–23667. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.E.; Siersema, P.D.; Vleggaar, F.P.; ten Kate, F.J.; Posthuma, G.; Souza, R.F.; de Haan, J.; van Baal, J.W.P.M. Toll-like receptor 2 signalling and the lysosomal machinery in Barretts esophagus. J. Gastrointest. Liver Dis. 2016, 25, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Tye, H.; Kennedy, C.L.; Najdovska, M.; McLeod, L.; McCormack, W.; Hughes, N.; Dev, A.; Sievert, W.; Ooi, C.H.; Ishikawa, T.O.; et al. STAT3-Driven Upregulation of TLR2 Promotes Gastric Tumorigenesis Independent of Tumor Inflammation. Cancer Cell 2012, 22, 466–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.; Anand, A.; Katarzyna, O.; Phelan, J.J.; Heeran, A.B.; Flis, E.; Clarke, N.E.; Watson, J.A.; Strangmann, J.; Flood, B.; et al. Characterizing caspase-1 involvement during esophageal disease progression. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pastuła, A.; Middelhoff, M.; Brandtner, A.; Tobiasch, M.; Höhl, B.; Nuber, A.H.; Demir, I.E.; Neupert, S.; Kollmann, P.; Mazzuoli-Weber, G.; et al. Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell Niche. Stem Cells Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, M.S.; Hussain, Z.; Giricz, O.; Shenoy, N.; Polineni, R.; Maitra, A.; Verma, A. Targeting Chemokine pathways in esophageal adenocarcinoma. Cell Cycle 2014, 13, 3320–3327. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesisin a dish: Modeling development and disease using organoid technologies. Science 2014, 345. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Quante, M.; Bhagat, G.; Abrams, J.; Marache, F.; Good, P.D.; Lee, M.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett’s-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [Green Version]
- Münch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hol, J.; Wilhelmsen, L.; Haraldsen, G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J. Leukoc. Biol. 2010, 87, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Lee, J.; Stamm, D.S.; Sanderson, I.R. MIP-2 secreted by epithelial cells increases neutrophil and lymphocyte recruitment in the mouse intestine. Gut 2001, 49, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, L.; Jiang, Y.; Song, L.; Liu, Y.; Cheng, F.; Fan, X.; Cao, X.; Gong, A.; Wang, D.; et al. Radiotherapy-induced cell death activates paracrine HMGB1-TLR2 signaling and accelerates pancreatic carcinoma metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef]
- Porter, R.J.; Murray, G.I.; Brice, D.P.; Petty, R.D.; McLean, M.H. Novel biomarkers for risk stratification of Barrett’s oesophagus associated neoplastic progression–epithelial HMGB1 expression and stromal lymphocytic phenotype. Br. J. Cancer 2019, 122, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.S.M.R.; Moriyama, M.; Kubota, K.; Ishiguro, N.; Sakamoto, M.; Chinju, A.; Mochizuki, K.; Sakamoto, T.; Kaneko, N.; Munemura, R.; et al. CD206+ tumor-associated macrophages promote proliferation and invasion in oral squamous cell carcinoma via EGF production. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 ininfection and immunity. Front. Immunol. 2012, 3, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.Y.; Nan, X.; Jin, M.S.; Youn, S.J.; Ryu, Y.H.; Mah, S.; Han, S.H.; Lee, H.; Paik, S.G.; Lee, J.O. Recognition of Lipopeptide Patterns by Toll-like Receptor 2-Toll-like Receptor 6 Heterodimer. Immunity 2009, 31, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Konturek, P.C.; Nikiforuk, A.; Kania, J.; Raithel, M.; Hahn, E.G.; Mühldorfer, S.M. Activation of NFκB represents the central event in the neoplastic progression associated with Barrett’s esophagus: A possible link to the inflammation and overexpression of COX-2, PPARγ and growth factors. Dig. Dis. Sci. 2004, 49, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C.; Abdalla, S.; Onwuegbusi, B.A.; Sirieix, P.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J.G. Inflammatory gradient in Barrett’s oesophagus: Implications for disease complications. Gut 2002, 51, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.M.; Splenser, A.E.; Kramer, J.; Fitzgerald, S.; Ramsey, D.; El-serag, H. Circulating Inflammatory Cytokines and Adipokines are Associated With Barrett’s Esophagus: A Case–Control Study. Clin. Gastroenterol. Hepatol. 2014, 12, 229–238. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Choi, H.J.; Ha, S.J.; Lee, K.T.; Kwon, Y.G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta 2013, 1835, 170–179. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.; Descargues, P.; Kim, Y.; Luo, J.; Karin, M. Carcinoma Produced Factors Activate Myeloid Cells via TLR2 to Stimulate Metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-Conditioned Macrophages Secrete Migration-Stimulating Factor: A New Marker for M2-Polarization, Influencing Tumor Cell Motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Peters, J.H.; Nieman, D.; Sharma, M.; Watson, T.; Yu, J. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br. J. Cancer 2015, 113, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- He, S.J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Song, T.; Wang, F.; Yin, C.; Li, Z.; Lin, J.; Meng, Y.; Feng, H. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1 + TAM expansion. Oncogenesis 2019, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Karin, M. Role of TLR2-dependent inflammation in metastatic progression. Ann. N. Y. Acad. Sci. 2011, 1217, 191–206. [Google Scholar] [CrossRef]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flis, E.; Barber, G.; Nulty, C.; Keogh, B.; McGuirk, P.; Anand, A.; O’Sullivan, J.; Quante, M.; Creagh, E.M. Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression. Cancers 2021, 13, 2065. https://doi.org/10.3390/cancers13092065
Flis E, Barber G, Nulty C, Keogh B, McGuirk P, Anand A, O’Sullivan J, Quante M, Creagh EM. Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression. Cancers. 2021; 13(9):2065. https://doi.org/10.3390/cancers13092065
Chicago/Turabian StyleFlis, Ewelina, Gillian Barber, Ciara Nulty, Brian Keogh, Peter McGuirk, Akanksha Anand, Jacintha O’Sullivan, Michael Quante, and Emma M. Creagh. 2021. "Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression" Cancers 13, no. 9: 2065. https://doi.org/10.3390/cancers13092065
APA StyleFlis, E., Barber, G., Nulty, C., Keogh, B., McGuirk, P., Anand, A., O’Sullivan, J., Quante, M., & Creagh, E. M. (2021). Identification of TLR2 Signalling Mechanisms Which Contribute to Barrett’s and Oesophageal Adenocarcinoma Disease Progression. Cancers, 13(9), 2065. https://doi.org/10.3390/cancers13092065