
Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms



Abstract
:Simple Summary
Abstract
1. Introduction
2. Diagnostic Criteria of MDS/MPN
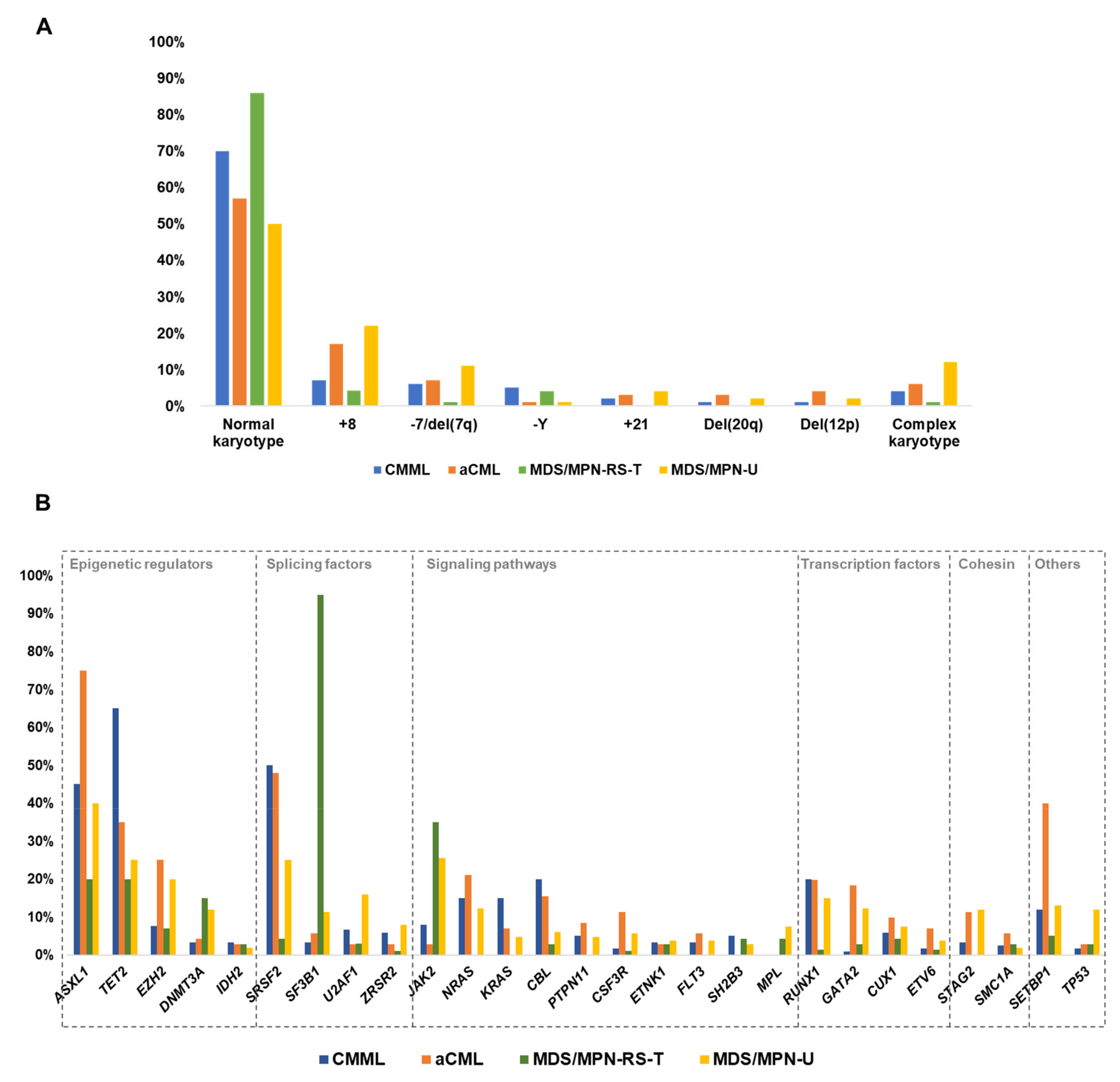
3. Cytogenetic Abnormalities in MDS/MPN
4. Other Chromosomal Abnormalities
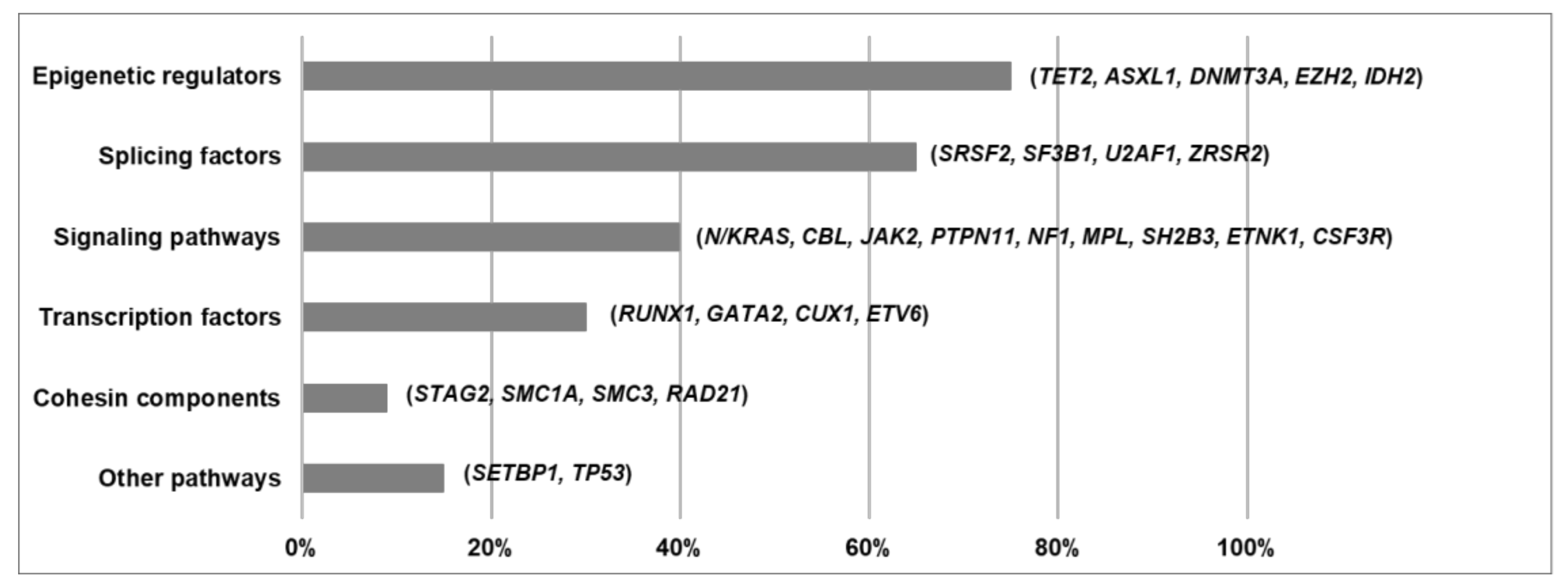
5. Functional Pathways Affected in MDS/MPN
5.1. Epigenetic Regulators
5.2. Splicing Factors
5.3. Signaling Pathways
5.4. Transcription Factors
5.5. Cohesin Components
5.6. Other Functional Pathways
6. Molecular Landscape of MDS/MPN and Clinical Implications
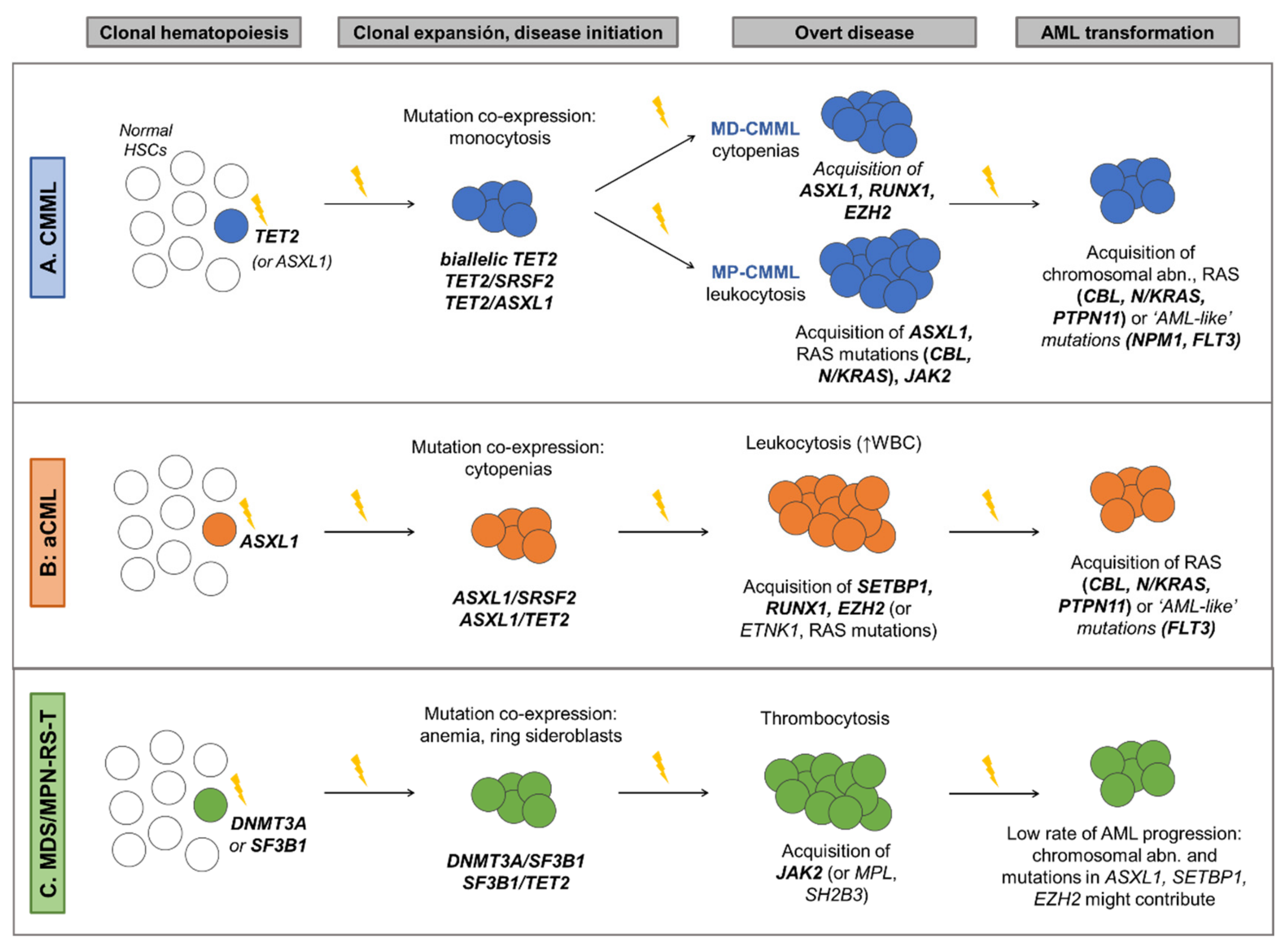
6.1. Chronic Myelomonocytic Leukemia
6.2. Atypical Chronic Myeloid Leukemia
6.3. MDS/MPN with Ring Sideroblasts and Thrombocytosis
6.4. MDS/MPN Unclassifiable
6.5. Juvenile Myelomonocytic Leukemia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JMML Subtype and Frequency (%) | Age of Onset (Years, Median) | Mutation (Type and Location) | Clinical Features | Prognosis and Treatment Implications |
|---|---|---|---|---|
| PTPN11 (40%) | 2.1 | Somatic missense mutations affecting exons 3 and 13. | Acquisition of NF1 haploinsufficiency is a frequent subclonal event. | Rapidly fatal unless allogenic HSCT can be successfully performed. |
| NRAS (18%) | 1.2 | Somatic missense mutations affecting exon 2. | • Subtype with the highest clinical diversity. • Clinically, patients are well and show a normal or slightly elevated HbF. | Although a considerable percentage relapse after HSCT, others survive in its absence and that of slowly regressing disease. |
| KRAS (14%) | 0.9 | Somatic missense mutations affecting exon 2. One half of cases present monosomy 7. | • Most children are diagnosed before the age of 1 year. • They often present with particularly severe disease. | Low relapse rate after allogeneic HSCT. |
| CBL (12–18%) | 0.9 | Germline mutations located throughout the linker and ring finger domain (intron 7, exons 8 and 9). Most patients have 11q isodisomy in hematopoietic cells. | Most children with CBL mutations have self-limiting disease with persistence of clonal hematopoiesis. | Observation without therapeutic intervention is generally advised. Value of allogenic HSCT is uncertain |
| NF1 (5%) | 2.8 | ≈65%: LOH at NF1 locus caused by UPD of 17q ≈35%: compound heterozygous NF1 inactivating mutations. Minority of cases: somatic interstitial deletions. | • Higher platelet count. • Higher percentage of bone marrow blasts. | Invariably fatal unless allogenic HSCT is successful. |
7. Practical Consideration within the Clinical Context
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Gelsi-Boyer, V.; Meggendorfer, M.; Morabito, M.; Berthon, C.; Adès, L.; Fenaux, P.; Beyne-Rauzy, O.; et al. Prognostic Score Including Gene Mutations in Chronic Myelomonocytic Leukemia. JCO 2013, 31, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Barraco, D.; Lasho, T.L.; Finke, C.M.; Reichard, K.; Hoversten, K.P.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Targeted next Generation Sequencing and Identification of Risk Factors in World Health Organization Defined Atypical Chronic Myeloid Leukemia. Am. J. Hematol. 2017, 92, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Ademà, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular Landscape and Clonal Architecture of Adult Myelodysplastic/Myeloproliferative Neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Rollison, D.E.; Howlader, N.; Smith, M.T.; Strom, S.S.; Merritt, W.D.; Ries, L.A.; Edwards, B.K.; List, A.F. Epidemiology of Myelodysplastic Syndromes and Chronic Myeloproliferative Disorders in the United States, 2001–2004, Using Data from the NAACCR and SEER Programs. Blood 2008, 112, 45–52. [Google Scholar] [CrossRef]
- Solary, E.; Itzykson, R. How I Treat Chronic Myelomonocytic Leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, M.M.; Tefferi, A. Chronic Myelomonocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Management. Am. J. Hematol. 2020, 95, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.; Sultan, C.; Cox, C. The Chronic Myeloid Leukaemias: Guidelines for Distinguishing Chronic Granulocytic, Atypical Chronic Myeloid, and Chronic Myelomonocytic Leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br. J. Haematol. 1994, 87, 746–754. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001. [Google Scholar]
- Such, E.; Germing, U.; Malcovati, L.; Cervera, J.; Kuendgen, A.; Della Porta, M.G.; Nomdedeu, B.; Arenillas, L.; Luño, E.; Xicoy, B.; et al. Development and Validation of a Prognostic Scoring System for Patients with Chronic Myelomonocytic Leukemia. Blood 2013, 121, 3005–3015. [Google Scholar] [CrossRef]
- Schuler, E.; Schroeder, M.; Neukirchen, J.; Strupp, C.; Xicoy, B.; Kündgen, A.; Hildebrandt, B.; Haas, R.; Gattermann, N.; Germing, U. Refined Medullary Blast and White Blood Cell Count Based Classification of Chronic Myelomonocytic Leukemias. Leuk. Res. 2014, 38, 1413–1419. [Google Scholar] [CrossRef]
- Orazi, A.; Germing, U. The Myelodysplastic/Myeloproliferative Neoplasms: Myeloproliferative Diseases with Dysplastic Features. Leukemia 2008, 22, 1308–1319. [Google Scholar] [CrossRef] [Green Version]
- Breccia, M.; Biondo, F.; Latagliata, R.; Carmosino, I.; Mandelli, F.; Alimena, G. Identification of Risk Factors in Atypical Chronic Myeloid Leukemia. Haematologica 2006, 91, 1566–1568. [Google Scholar]
- Giri, S.; Pathak, R.; Martin, M.G.; Bhatt, V.R. Characteristics and Survival of BCR/ABL Negative Chronic Myeloid Leukemia: A Retrospective Analysis of the Surveillance, Epidemiology and End Results Database. Ther. Adv. Hematol. 2015, 6, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Jeromin, S.; Haferlach, T.; Weissmann, S.; Meggendorfer, M.; Eder, C.; Nadarajah, N.; Alpermann, T.; Kohlmann, A.; Kern, W.; Haferlach, C.; et al. Refractory Anemia with Ring Sideroblasts and Marked Thrombocytosis Cases Harbor Mutations in SF3B1 or Other Spliceosome Genes Accompanied by JAK2V617F and ASXL1 Mutations. Haematologica 2015, 100, e125–e127. [Google Scholar] [CrossRef]
- Broseus, J.; Florensa, L.; Zipperer, E.; Schnittger, S.; Malcovati, L.; Richebourg, S.; Lippert, E.; Cermak, J.; Evans, J.; Mounier, M.; et al. Clinical Features and Course of Refractory Anemia with Ring Sideroblasts Associated with Marked Thrombocytosis. Haematologica 2012, 97, 1036–1041. [Google Scholar] [CrossRef]
- Atallah, E.; Nussenzveig, R.; Yin, C.C.; Bueso-Ramos, C.; Tam, C.; Manshouri, T.; Pierce, S.; Kantarjian, H.; Verstovsek, S. Prognostic Interaction between Thrombocytosis and JAK2 V617F Mutation in the WHO Subcategories of Myelodysplastic/Myeloproliferative Disease-Unclassifiable and Refractory Anemia with Ringed Sideroblasts and Marked Thrombocytosis. Leukemia 2008, 22, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Cannella, L.; Breccia, M.; Latagliata, R.; Frustaci, A.; Alimena, G. Clinical and Prognostic Features of Patients with Myelodysplastic/Myeloproliferative Syndrome Categorized as Unclassified (MDS/MPD-U) by WHO Classification. Leuk. Res. 2008, 32, 514–516. [Google Scholar] [CrossRef]
- Wang, S.A.; Hasserjian, R.P.; Fox, P.S.; Rogers, H.J.; Geyer, J.T.; Chabot-Richards, D.; Weinzierl, E.; Hatem, J.; Jaso, J.; Kanagal-Shamanna, R.; et al. Atypical Chronic Myeloid Leukemia Is Clinically Distinct from Unclassifiable Myelodysplastic/Myeloproliferative Neoplasms. Blood 2014, 123, 2645–2651. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Daver, N.; Jain, N.; Pemmaraju, N.; Bueso-Ramos, C.; Yin, C.C.; Pierce, S.; Jabbour, E.; Cortes, J.E.; Kantarjian, H.M.; et al. Myelodysplastic/Myeloproliferative Neoplasms, Unclassifiable (MDS/MPN, U): Natural History and Clinical Outcome by Treatment Strategy. Leukemia 2014, 28, 958–961. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Arico, M.; Basso, G.; Biondi, A.; Cantu Rajnoldi, A.; Creutzig, U.; Haas, O.; Harbott, J.; Hasle, H.; Kerndrup, G.; et al. Chronic Myelomonocytic Leukemia in Childhood: A Retrospective Analysis of 110 Cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997, 89, 3534–3543. [Google Scholar]
- Niemeyer, C.M.; Flotho, C. Juvenile Myelomonocytic Leukemia: Who’s the Driver at the Wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Such, E.; Cervera, J.; Costa, D.; Sole, F.; Vallespi, T.; Luno, E.; Collado, R.; Calasanz, M.J.; Hernandez-Rivas, J.M.; Cigudosa, J.C.; et al. Cytogenetic Risk Stratification in Chronic Myelomonocytic Leukemia. Haematologica 2011, 96, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Zhang, L.; Fu, B.; Hu, J.; Lu, X.; Hu, S.; Patel, A.; Goswami, M.; Khoury, J.D.; Garcia-Manero, G.; et al. Cytogenetic Risk Stratification of 417 Patients with Chronic Myelomonocytic Leukemia from a Single Institution: Cytogenetic Stratification in CMML Patients. Am. J. Hematol. 2014, 89, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassie, E.A.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Solary, E.; Tefferi, A.; Patnaik, M.M. Molecular and Prognostic Correlates of Cytogenetic Abnormalities in Chronic Myelomonocytic Leukemia: A Mayo Clinic-French Consortium Study. Am. J. Hematol. 2014, 89, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Predictors of Survival in Refractory Anemia with Ring Sideroblasts and Thrombocytosis (RARS-T) and the Role of next-Generation Sequencing: Predictors of Survival in Refractory Anemia with Ring Sideroblasts and Thrombocytosis. Am. J. Hematol. 2016, 91, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangaonkar, A.A.; Swoboda, D.M.; Coltro, G.; Lasho, T.L.; Novotny, P.J.; Pophali, P.; Carr, R.M.; Binder, M.; Finke, C.M.; Gangat, N.; et al. Clinicopathologic Characteristics, Prognostication and Treatment Outcomes for Myelodysplastic/Myeloproliferative Neoplasm, Unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center Study of 135 Consecutive Patients. Leukemia 2020, 34, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Pierre, R.V.; Hoagland, H.C. Age-Associated Aneuploidy: Loss of Y Chromosome from Human Bone Marrow Cells with Aging. Cancer 1972, 30, 889–894. [Google Scholar] [CrossRef]
- Guttenbach, M.; Koschorz, B.; Bernthaler, U.; Grimm, T.; Schmid, M. Sex Chromosome Loss and Aging: In Situ Hybridization Studies on Human Interphase Nuclei. Am. J. Hum. Genet. 1995, 57, 1143. [Google Scholar]
- Elena, C.; Gallì, A.; Such, E.; Meggendorfer, M.; Germing, U.; Rizzo, E.; Cervera, J.; Molteni, E.; Fasan, A.; Schuler, E.; et al. Integrating Clinical Features and Genetic Lesions in the Risk Assessment of Patients with Chronic Myelomonocytic Leukemia. Blood 2016, 128, 1408–1417. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Knudson, R.A.; Ketterling, R.P.; Tefferi, A.; Solary, E. ASXL1 and SETBP1 Mutations and Their Prognostic Contribution in Chronic Myelomonocytic Leukemia: A Two-Center Study of 466 Patients. Leukemia 2014, 28, 2206–2212. [Google Scholar] [CrossRef]
- Coltro, G.; Mangaonkar, A.A.; Lasho, T.L.; Finke, C.M.; Pophali, P.; Carr, R.; Gangat, N.; Binder, M.; Pardanani, A.; Fernandez-Zapico, M.; et al. Clinical, Molecular, and Prognostic Correlates of Number, Type, and Functional Localization of TET2 Mutations in Chronic Myelomonocytic Leukemia (CMML)-a Study of 1084 Patients. Leukemia 2020, 34, 1407–1421. [Google Scholar] [CrossRef]
- Palomo, L.; Garcia, O.; Arnan, M.; Xicoy, B.; Fuster, F.; Cabezón, M.; Coll, R.; Ademà, V.; Grau, J.; Jiménez, M.-J.; et al. Targeted Deep Sequencing Improves Outcome Stratification in Chronic Myelomonocytic Leukemia with Low Risk Cytogenetic Features. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Vijayvargiya, P.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Prognostic Interaction between ASXL1 and TET2 Mutations in Chronic Myelomonocytic Leukemia. Blood Cancer J. 2016, 6, e385. [Google Scholar] [CrossRef] [Green Version]
- Meggendorfer, M.; Bacher, U.; Alpermann, T.; Haferlach, C.; Kern, W.; Gambacorti-Passerini, C.; Haferlach, T.; Schnittger, S. SETBP1 Mutations Occur in 9% of MDS/MPN and in 4% of MPN Cases and Are Strongly Associated with Atypical CML, Monosomy 7, Isochromosome i(17)(Q10), ASXL1 and CBL Mutations. Leukemia 2013, 27, 1852–1860. [Google Scholar] [CrossRef]
- Broséus, J.; Lippert, E.; Harutyunyan, A.S.; Jeromin, S.; Zipperer, E.; Florensa, L.; Milosevic, J.D.; Haferlach, T.; Germing, U.; Luño, E.; et al. Low Rate of Calreticulin Mutations in Refractory Anaemia with Ring Sideroblasts and Marked Thrombocytosis. Leukemia 2014, 28, 1374–1376. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Nazha, A.; Komrokji, R.S.; Patel, K.P.; Pierce, S.A.; Al-Ali, N.; Sochacki, A.; Shaver, A.; Ma, W.; Su, X.; et al. Mutational Landscape of Myelodysplastic/Myeloproliferative Neoplasm-Unclassifiable. Blood 2018, 132, 2100–2103. [Google Scholar] [CrossRef] [Green Version]
- Lipka, D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.; Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-Pathway Mutation Patterns Define Epigenetic Subclasses in Juvenile Myelomonocytic Leukemia. Nat. Commun. 2017, 8, 2126. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, H.; Okuno, Y.; Muramatsu, H.; Yoshida, K.; Shiraishi, Y.; Takahashi, M.; Kon, A.; Sanada, M.; Chiba, K.; Tanaka, H.; et al. Exome Sequencing Identifies Secondary Mutations of SETBP1 and JAK3 in Juvenile Myelomonocytic Leukemia. Nat. Genet. 2013, 45, 937–941. [Google Scholar] [CrossRef]
- Murakami, N.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Nagae, G.; Suzuki, K.; Narita, A.; Sakaguchi, H.; Kawashima, N.; Wang, X.; et al. Integrated Molecular Profiling of Juvenile Myelomonocytic Leukemia. Blood 2018, 131, 1576–1586. [Google Scholar] [CrossRef]
- Gondek, L.P.; Dunbar, A.J.; Szpurka, H.; McDevitt, M.A.; Maciejewski, J.P. SNP Array Karyotyping Allows for the Detection of Uniparental Disomy and Cryptic Chromosomal Abnormalities in MDS/MPD-U and MPD. PLoS ONE 2007, 2, e1225. [Google Scholar] [CrossRef]
- Gondek, L.P.; Tiu, R.; O’Keefe, C.L.; Sekeres, M.A.; Theil, K.S.; Maciejewski, J.P. Chromosomal Lesions and Uniparental Disomy Detected by SNP Arrays in MDS, MDS/MPD, and MDS-Derived AML. Blood 2008, 111, 1534–1542. [Google Scholar] [CrossRef] [Green Version]
- Tiu, R.V.; Gondek, L.P.; O’Keefe, C.L.; Elson, P.; Huh, J.; Mohamedali, A.; Kulasekararaj, A.; Advani, A.S.; Paquette, R.; List, A.F.; et al. Prognostic Impact of SNP Array Karyotyping in Myelodysplastic Syndromes and Related Myeloid Malignancies. Blood 2011, 117, 4552–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, A.J.; Gondek, L.P.; O’Keefe, C.L.; Makishima, H.; Rataul, M.S.; Szpurka, H.; Sekeres, M.A.; Wang, X.F.; McDevitt, M.A.; Maciejewski, J.P. 250K Single Nucleotide Polymorphism Array Karyotyping Identifies Acquired Uniparental Disomy and Homozygous Mutations, Including Novel Missense Substitutions of c-Cbl, in Myeloid Malignancies. Cancer Res. 2008, 68, 10349–10357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, L.; Xicoy, B.; Garcia, O.; Mallo, M.; Ademà, V.; Cabezón, M.; Arnan, M.; Pomares, H.; José Larrayoz, M.; José Calasanz, M.; et al. Impact of SNP Array Karyotyping on the Diagnosis and the Outcome of Chronic Myelomonocytic Leukemia with Low Risk Cytogenetic Features or No Metaphases. Am. J. Hematol. 2016, 91, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetro, C.; Haferlach, C.; Haferlach, T.; Zenger, M.; Nadarajah, N.; Kern, W.; Meggendorfer, M. Aberrations Identified by Genomic Arrays in Normal Karyotype CMML Can Be Detected in 40% of Patients, but Do Not Add Prognostic Information to Molecular Mutations. Leukemia 2016, 30, 2235–2238. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of Heterozygosity 4q24 and TET2 Mutations Associated with Myelodysplastic/Myeloproliferative Neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagal-Shamanna, R.; Hodge, J.C.; Tucker, T.; Shetty, S.; Yenamandra, A.; Dixon-McIver, A.; Bryke, C.; Huxley, E.; Lennon, P.A.; Raca, G.; et al. Assessing Copy Number Aberrations and Copy Neutral Loss of Heterozygosity across the Genome as Best Practice: An Evidence Based Review of Clinical Utility from the Cancer Genomics Consortium (CGC) Working Group for Myelodysplastic Syndrome, Myelodysplastic/Myeloproliferative and Myeloproliferative Neoplasms. Cancer Genet. 2018, 228–229, 197–217. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Morabito, M.; Preudhomme, C.; Berthon, C.; Adès, L.; Fenaux, P.; Platzbecker, U.; Gagey, O.; et al. Clonal Architecture of Chronic Myelomonocytic Leukemias. Blood 2013, 121, 2186–2198. [Google Scholar] [CrossRef]
- Ricci, C.; Fermo, E.; Corti, S.; Molteni, M.; Faricciotti, A.; Cortelezzi, A.; Lambertenghi Deliliers, G.; Beran, M.; Onida, F. RAS Mutations Contribute to Evolution of Chronic Myelomonocytic Leukemia to the Proliferative Variant. Clin. Cancer Res. 2010, 16, 2246–2256. [Google Scholar] [CrossRef] [Green Version]
- Mason, C.C.; Khorashad, J.S.; Tantravahi, S.K.; Kelley, T.W.; Zabriskie, M.S.; Yan, D.; Pomicter, A.D.; Reynolds, K.R.; Eiring, A.M.; Kronenberg, Z.; et al. Age-Related Mutations and Chronic Myelomonocytic Leukemia. Leukemia 2016, 30, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Awada, H.; Nagata, Y.; Goyal, A.; Asad, M.F.; Patel, B.; Hirsch, C.M.; Kuzmanovic, T.; Guan, Y.; Przychodzen, B.P.; Aly, M.; et al. Invariant Phenotype and Molecular Association of Biallelic TET2 Mutant Myeloid Neoplasia. Blood Adv. 2019, 3, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.J.; Przychodzen, B.; Thota, S.; Radivoyevitch, T.; Visconte, V.; Kuzmanovic, T.; Clemente, M.; Hirsch, C.; Morawski, A.; Souaid, R.; et al. Genomic Determinants of Chronic Myelomonocytic Leukemia. Leukemia 2017, 31, 2815–2823. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The Role of Mutations in Epigenetic Regulators in Myeloid Malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired Hydroxylation of 5-Methylcytosine in Myeloid Cancers with Mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Damm, F.; Itzykson, R.; Kosmider, O.; Droin, N.; Renneville, A.; Chesnais, V.; Gelsi-Boyer, V.; de Botton, S.; Vey, N.; Preudhomme, C.; et al. SETBP1 Mutations in 658 Patients with Myelodysplastic Syndromes, Chronic Myelomonocytic Leukemia and Secondary Acute Myeloid Leukemias. Leukemia 2013, 27, 1401–1403. [Google Scholar] [CrossRef]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A Mutations in Patients with Myelodysplastic Syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 Dominate Clonal Hematopoiesis and Demonstrate Benign Phenotypes and Different Genetic Predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Zhao, H.; Hardikar, S.; Singh, A.K.; Goodell, M.A.; Chen, T. A DNMT3A Mutation Common in AML Exhibits Dominant-Negative Effects in Murine ES Cells. Blood 2013, 122, 4086–4089. [Google Scholar] [CrossRef] [Green Version]
- Martín, I.; Such, E.; Navarro, B.; Vicente, A.; López-Pavía, M.; Ibáñez, M.; Tormo, M.; Villamón, E.; Gómez-Seguí, I.; Luna, I.; et al. Negative Impact on Clinical Outcome of the Mutational Co-Occurrence of SF3B1 and DNMT3A in Refractory Anemia with Ring Sideroblasts (RARS). Leuk Lymphoma 2017, 58, 1686–1693. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.-J.; Birnbaum, D. Mutations in ASXL1 Are Associated with Poor Prognosis across the Spectrum of Malignant Myeloid Diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of Heterozygosity in 7q Myeloid Disorders: Clinical Associations and Genomic Pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the Spliceosome Machinery, a Novel and Ubiquitous Pathway in Leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broséus, J.; Alpermann, T.; Wulfert, M.; Florensa Brichs, L.; Jeromin, S.; Lippert, E.; Rozman, M.; Lifermann, F.; Grossmann, V.; Haferlach, T.; et al. Age, JAK2(V617F) and SF3B1 Mutations Are the Main Predicting Factors for Survival in Refractory Anaemia with Ring Sideroblasts and Marked Thrombocytosis. Leukemia 2013, 27, 1826–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant Splicing of U12-Type Introns Is the Hallmark of ZRSR2 Mutant Myelodysplastic Syndrome. Nat. Commun 2015, 6, 6042. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-MRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The Genetic Basis of Myelodysplasia and Its Clinical Relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Kon, A.; Shih, L.-Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent Mutations in Multiple Components of the Cohesin Complex in Myeloid Neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 Mutations in Atypical Chronic Myeloid Leukemia. Nat. Genet. 2013, 45, 18–24. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshida, K.; Nguyen, N.; Przychodzen, B.; Sanada, M.; Okuno, Y.; Ng, K.P.; Gudmundsson, K.O.; Vishwakarma, B.A.; Jerez, A.; et al. Somatic SETBP1 Mutations in Myeloid Malignancies. Nat. Genet. 2013, 45, 942–946. [Google Scholar] [CrossRef]
- Hou, H.-A.; Kuo, Y.-Y.; Tang, J.-L.; Chou, W.-C.; Yao, M.; Lai, Y.-J.; Lin, C.-C.; Chen, C.-Y.; Liu, C.-Y.; Tseng, M.-H.; et al. Clinical Implications of the SETBP1 Mutation in Patients with Primary Myelodysplastic Syndrome and Its Stability during Disease Progression. Am. J. Hematol. 2014, 89, 181–186. [Google Scholar] [CrossRef]
- Adema, V.; Larráyoz, M.J.; Calasanz, M.J.; Palomo, L.; Patiño-García, A.; Agirre, X.; Hernández-Rivas, J.M.; Lumbreras, E.; Buño, I.; Martinez-Laperche, C.; et al. Correlation of Myelodysplastic Syndromes with i(17)(Q10) and TP53 and SETBP1 Mutations. Br. J. Haematol. 2015, 171, 137–141. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Vallapureddy, R.; Yalniz, F.F.; Hanson, C.A.; Ketterling, R.P.; Lasho, T.L.; Finke, C.; Al-Kali, A.; Gangat, N.; Tefferi, A. Therapy Related-Chronic Myelomonocytic Leukemia (CMML): Molecular, Cytogenetic, and Clinical Distinctions from de Novo CMML. Am. J. Hematol 2018, 93, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Shih, L.-Y.; Lin, T.-L.; Wang, P.-N.; Wu, J.-H.; Dunn, P.; Kuo, M.-C.; Huang, C.-F. Internal Tandem Duplication of Fms-like Tyrosine Kinase 3 Is Associated with Poor Outcome in Patients with Myelodysplastic Syndrome. Cancer 2004, 101, 989–998. [Google Scholar] [CrossRef]
- Vallapureddy, R.; Lasho, T.L.; Hoversten, K.; Finke, C.M.; Ketterling, R.; Hanson, C.; Gangat, N.; Tefferi, A.; Patnaik, M.M. Nucleophosmin 1 (NPM1) Mutations in Chronic Myelomonocytic Leukemia and Their Prognostic Relevance. Am. J. Hematol. 2017, 92, E614–E618. [Google Scholar] [CrossRef] [Green Version]
- Merlevede, J.; Droin, N.; Qin, T.; Meldi, K.; Yoshida, K.; Morabito, M.; Chautard, E.; Auboeuf, D.; Fenaux, P.; Braun, T.; et al. Mutation Allele Burden Remains Unchanged in Chronic Myelomonocytic Leukaemia Responding to Hypomethylating Agents. Nat. Commun. 2016, 7, 10767. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Maxson, J.E.; George, T.I.; Tyner, J.W. The New Genetics of Chronic Neutrophilic Leukemia and Atypical CML: Implications for Diagnosis and Treatment. Blood 2013, 122, 1707–1711. [Google Scholar] [CrossRef] [Green Version]
- Crisà, E.; Nicolosi, M.; Ferri, V.; Favini, C.; Gaidano, G.; Patriarca, A. Atypical Chronic Myeloid Leukemia: Where Are We Now? Int. J. Mol. Sci. 2020, 21, 6862. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; King, R.L.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Vascular Events and Risk Factors for Thrombosis in Refractory Anemia with Ring Sideroblasts and Thrombocytosis. Leukemia 2016, 30, 2273–2275. [Google Scholar] [CrossRef]
- Calvo, X.; Garcia-Gisbert, N.; Parraga, I.; Gibert, J.; Florensa, L.; Andrade-Campos, M.; Merchan, B.; Garcia-Avila, S.; Montesdeoca, S.; Fernández-Rodríguez, C.; et al. Oligomonocytic and Overt Chronic Myelomonocytic Leukemia Show Similar Clinical, Genomic, and Immunophenotypic Features. Blood Adv. 2020, 4, 5285–5296. [Google Scholar] [CrossRef]
- Flex, E.; Jaiswal, M.; Pantaleoni, F.; Martinelli, S.; Strullu, M.; Fansa, E.K.; Caye, A.; De Luca, A.; Lepri, F.; Dvorsky, R.; et al. Activating Mutations in RRAS Underlie a Phenotype within the RASopathy Spectrum and Contribute to Leukaemogenesis. Hum. Mol. Genet. 2014, 23, 4315–4327. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M. JMML Genomics and Decisions. Hematology 2018, 2018, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.-D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.R.; et al. The Genomic Landscape of Juvenile Myelomonocytic Leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Muramatsu, H.; Makishima, H.; Prince, C.; Jankowska, A.M.; Yoshida, N.; Xu, Y.; Nishio, N.; Hama, A.; Yagasaki, H.; et al. Spectrum of Molecular Defects in Juvenile Myelomonocytic Leukaemia Includes ASXL1 Mutations. Br. J. Haematol. 2010, 150, 83–87. [Google Scholar] [CrossRef]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile Myelomonocytic Leukemia Displays Mutations in Components of the RAS Pathway and the PRC2 Network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Bresolin, S.; De Filippi, P.; Vendemini, F.; D’Alia, M.; Zecca, M.; Meyer, L.H.; Danesino, C.; Locatelli, F.; Masetti, R.; Basso, G.; et al. Mutations of SETBP1 and JAK3 in Juvenile Myelomonocytic Leukemia: A Report from the Italian AIEOP Study Group. Oncotarget 2016, 7, 28914–28919. [Google Scholar] [CrossRef]
- Stieglitz, E.; Troup, C.B.; Gelston, L.C.; Haliburton, J.; Chow, E.D.; Yu, K.B.; Akutagawa, J.; Taylor-Weiner, A.N.; Liu, Y.L.; Wang, Y.-D.; et al. Subclonal Mutations in SETBP1 Confer a Poor Prognosis in Juvenile Myelomonocytic Leukemia. Blood 2015, 125, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Mazor, T.; Olshen, A.B.; Geng, H.; Gelston, L.C.; Akutagawa, J.; Lipka, D.B.; Plass, C.; Flotho, C.; Chehab, F.F.; et al. Genome-Wide DNA Methylation Is Predictive of Outcome in Juvenile Myelomonocytic Leukemia. Nat. Commun. 2017, 8, 2127. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Schroeder, T.; Zabelina, T.; Badbaran, A.; Bacher, U.; Kobbe, G.; Ayuk, F.; Wolschke, C.; Schnittger, S.; Kohlmann, A.; et al. Postallogeneic Monitoring with Molecular Markers Detected by Pretransplant Next-Generation or Sanger Sequencing Predicts Clinical Relapse in Patients with Myelodysplastic/Myeloproliferative Neoplasms. Eur. J. Haematol. 2014, 92, 189–194. [Google Scholar] [CrossRef]
- Carbonell, D.; Suárez-González, J.; Chicano, M.; Andrés-Zayas, C.; Triviño, J.C.; Rodríguez-Macías, G.; Bastos-Oreiro, M.; Font, P.; Ballesteros, M.; Muñiz, P.; et al. Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers 2019, 11, 1364. [Google Scholar] [CrossRef] [Green Version]
- Vantyghem, S.; Peterlin, P.; Thépot, S.; Ménard, A.; Dubruille, V.; Debord, C.; Guillaume, T.; Garnier, A.; Bourgeois, A.L.; Wuilleme, S.; et al. Diagnosis and prognosis are supported by integrated assessment of next-generation sequencing in chronic myeloid malignancies. A real-life study. Haematologica 2021, 106, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, Y.; Zhao, R.; Awada, H.; Kerr, C.M.; Mirzaev, I.; Kongkiatkamon, S.; Nazha, A.; Makishima, H.; Radivoyevitch, T.; Scott, J.G.; et al. Machine Learning Demonstrates That Somatic Mutations Imprint Invariant Morphologic Features in Myelodysplastic Syndromes. Blood 2020, 136, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T. Human and Artificial Intelligence to Illuminate MDS. Blood 2020, 136, 2243–2244. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Pfarr, N.; Meggendorfer, M.; Jost, P.; Haferlach, T.; Weichert, W. Next-generation diagnostics for precision oncology: Preanalytical considerations, technical challenges, and available technologies. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Pophali, P.; Matin, A.; Mangaonkar, A.A.; Carr, R.; Binder, M.; Al-Kali, A.; Begna, K.H.; Reichard, K.K.; Alkhateeb, H.; Shah, M.V.; et al. Prognostic impact and timing considerations for allogeneic hematopoietic stem cell transplantation in chronic myelomonocytic leukemia. Blood Cancer J. 2020, 10, 121. [Google Scholar] [CrossRef]
- Thota, S.; Gerds, A.T. Myelodysplastic and myeloproliferative neoplasms: Updates on the overlap syndromes. Leuk. Lymphoma 2018, 59, 803–812. [Google Scholar] [CrossRef]





| MDS/MPN | Abnormal Karyotypes (%) | Common Abnormalities (Frequency %) |
|---|---|---|
| CMML | 30% | +8: 6–7% −Y: 4–6% −7/del(7q): 2–9% +21: 1–2% CK: 3–6% Deletions of 20q (1–2%) and 12p (1%) |
| aCML | 43% | +8: 17% −7/del(7q): 6–8% CK: 4–8% |
| MDS/MPN-RS-T | 10–17% | +8: 4% −Y: 4% CK: 0–4% |
| MDS/MPN-U | 50% | +8: 14–25% −7/del(7q): 11% CK: 12% |
| JMML | 19–35% | −7: 9–25% Others (del(7q), +8): 10% |
| MDS/MPN Subtype | Diagnosis | Prognosis |
|---|---|---|
| CMML | -WHO [1]: presence of mutations in genes often associated with CMML (TET2, SRSF2, ASXL1, SETBP1) in the proper clinical contest can be used to support diagnosis -Associated with the following gene mutation combinations: TET2-SRSF2, biallelic TET2, SRSF2-RUNX1 [2,4,30] | Cytogenetics -Three cytogenetic stratification systems have been proposed [23,24,25] -Recurrent findings: • Low risk karyotypes: normal karyotype, isolated loss of Y • High risk karyotypes: chr7 abnormalities, complex karyotype, monosomal karyotype Gene mutations: -Unfavorable outcome: mutations in ASXL1, RUNX1, NRAS and SETBP1 [2,30,31] -Favorable outcome: TET2 mutations, especially in the absence of ASXL1 mutations (TET2MUT/ASXL1WT). These patients also show better response to HMA [32,33,34]. Prognostic stratification: -GFM Model [2], stratification in 3 risk groups based on: Age > 65 years; Hb < 10 g/dL in females and <11 g/dL in males; WBC > 15 × 109/L; Platelet count < 100 × 109/L; ASXL1 mutations -Mayo Molecular Model (MMM) [31], stratification in 4 risk groups based on: Hb < 10 g/dL; AMC > 10 × 109/L; Platelet count < 100 × 109/L; Presence of circulating IMCs; ASXL1 mutations -CPSS-Mol [30], stratification in 4 risk groups based on: WBC ≥ 13 × 109; BM blasts ≥ 5%; RBC transfusion dependency; Genetic risk group (includes CMML-specific cytogenetic risk stratification [23] and mutations in ASXL1, RUNX1, NRAS and SETBP1). |
| aCML | -Associated with the following gene mutation combinations: ASXL1/SETBP1, SETBP1/SRSF2, ASXL1/EZH2, RUNX1/EZH2 [3,4,35] | Unfavorable outcome: mutations in TET2, RUNX1, NRAS and CUX1 [3,4] Prognostic stratification: Mayo Prognostic Model for aCML [3], stratification in 2 risk groups based on: Age > 67 years; Hb < 10 g/dL; TET2 mutations |
| MDS/MPN-RS-T | -WHO [1]: presence of a SF3B1 mutation. -Associated with the following gene mutation combinations: SF3B1, either alone or in combination with DNMT3A or JAK2, or DNMT3A/JAK2 [4,26,36] | Unfavorable outcome: -Presence of altered karyotype [4,26] -Mutations in ASXL1, SETBP1, EZH2 [4,26] Prognostic stratification: Mayo Prognostic Model for MDS/MPN-RS-T [26], stratification in 3 risk groups based on: Hb < 10 g/dL; Abnormal karyotype; mutations in ASXL1 or SETBP1 |
| MDS/MPN-U | - | Unfavorable outcome: -Presence of chr7 abnormalities and complex karyotypes [19] -Mutations in ASXL1, CBL, CEBPA, EZH2, STAG2, TP53 [4,27,37] Prognostic stratification: -Genomics-based stratification system (Figure 4), classification in 5 subtypes with prognostic relevance based on mutational profile [4] |
| JMML | -WHO [1]: presence of (1 finding sufficient): • Somatic mutation: PTPN11, KRAS, NRAS • Clinical diagnosis of NF1 or NF1 mutation • Germline CBL mutation CBL LOH | Prognostic stratification: According to the methylation level, three groups that correlate molecular features and clinical outcome have been proposed [38]: • High: characterized by somatic PTPN11 mutations and poor clinical outcome • Intermediate: enriched in somatic KRAS mutations and monosomy 7 • Low: characterized by somatic NRAS and CBL mutations and a favorable prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomo, L.; Acha, P.; Solé, F. Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms. Cancers 2021, 13, 2120. https://doi.org/10.3390/cancers13092120
Palomo L, Acha P, Solé F. Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms. Cancers. 2021; 13(9):2120. https://doi.org/10.3390/cancers13092120
Chicago/Turabian StylePalomo, Laura, Pamela Acha, and Francesc Solé. 2021. "Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms" Cancers 13, no. 9: 2120. https://doi.org/10.3390/cancers13092120
APA StylePalomo, L., Acha, P., & Solé, F. (2021). Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms. Cancers, 13(9), 2120. https://doi.org/10.3390/cancers13092120