
Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
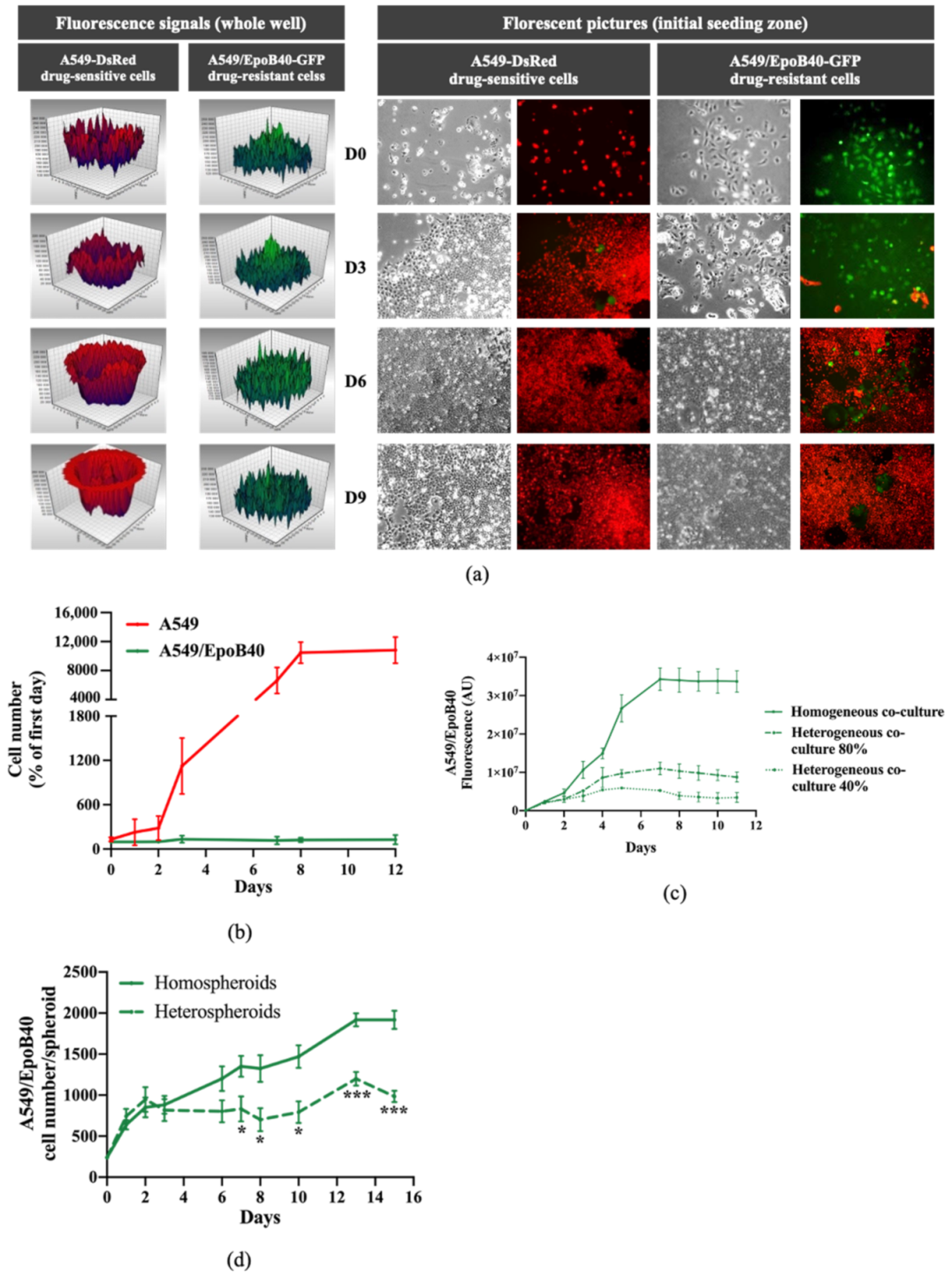
2.1. The Co-Culture System Demonstrates a Balance between Drug-Sensitive and Drug-Resistance Clones
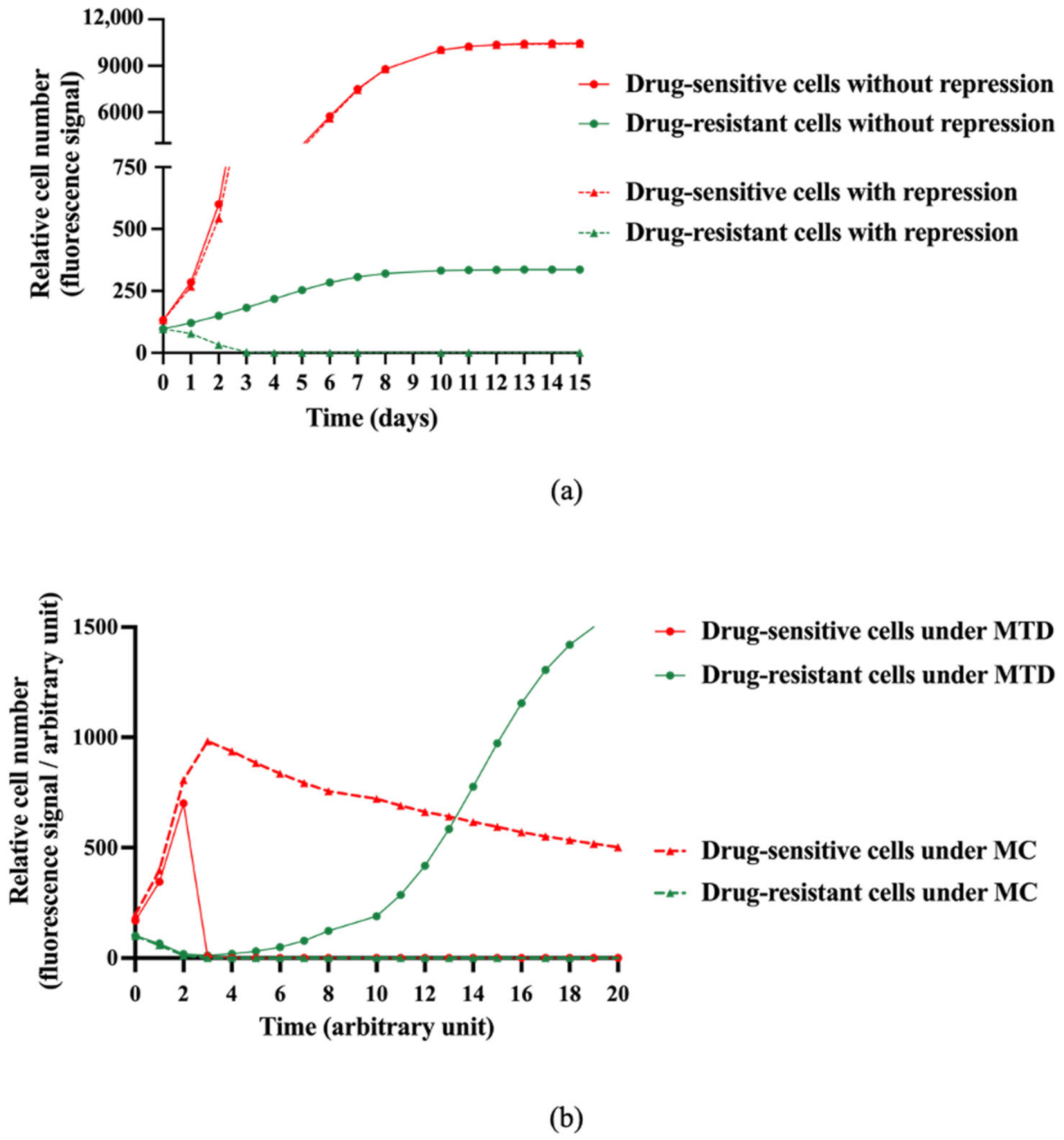
2.2. Mathematical Modeling Predicts that Metronomic Treatment Could Better Manage Intratumor Heterogeneity than the MTD Schedule
- In the well, there may be two types of cells, denoted respectively as drug-sensitive and drug-resistant cells.
- In the absence of the other cell type, drug-sensitive and -resistant cells can grow freely until the well becomes confluent.
- All cells from both types are competing to colonize the available space in the well.
- The presence of drug-sensitive cells in the well may have a suppressive role on the drug-resistant clone proliferation.
- The chemotherapeutic agent only acts on drug-sensitive cells.
- A delay between the injection of the chemotherapeutic agent and its impact on cell functions is taken into account.
- The chemotherapeutic agent concentration is assumed constant in the absence of an experimenter intervention.
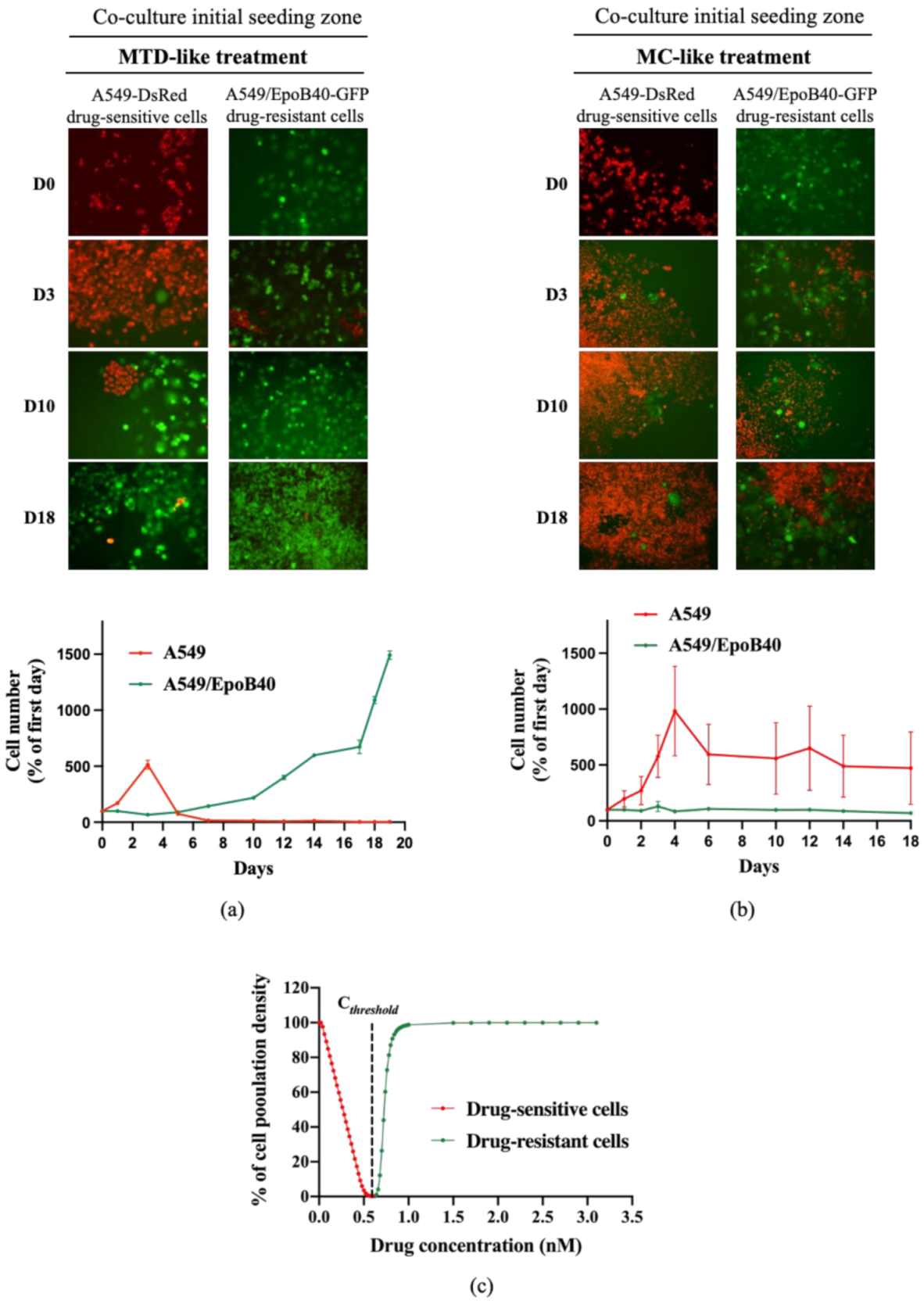
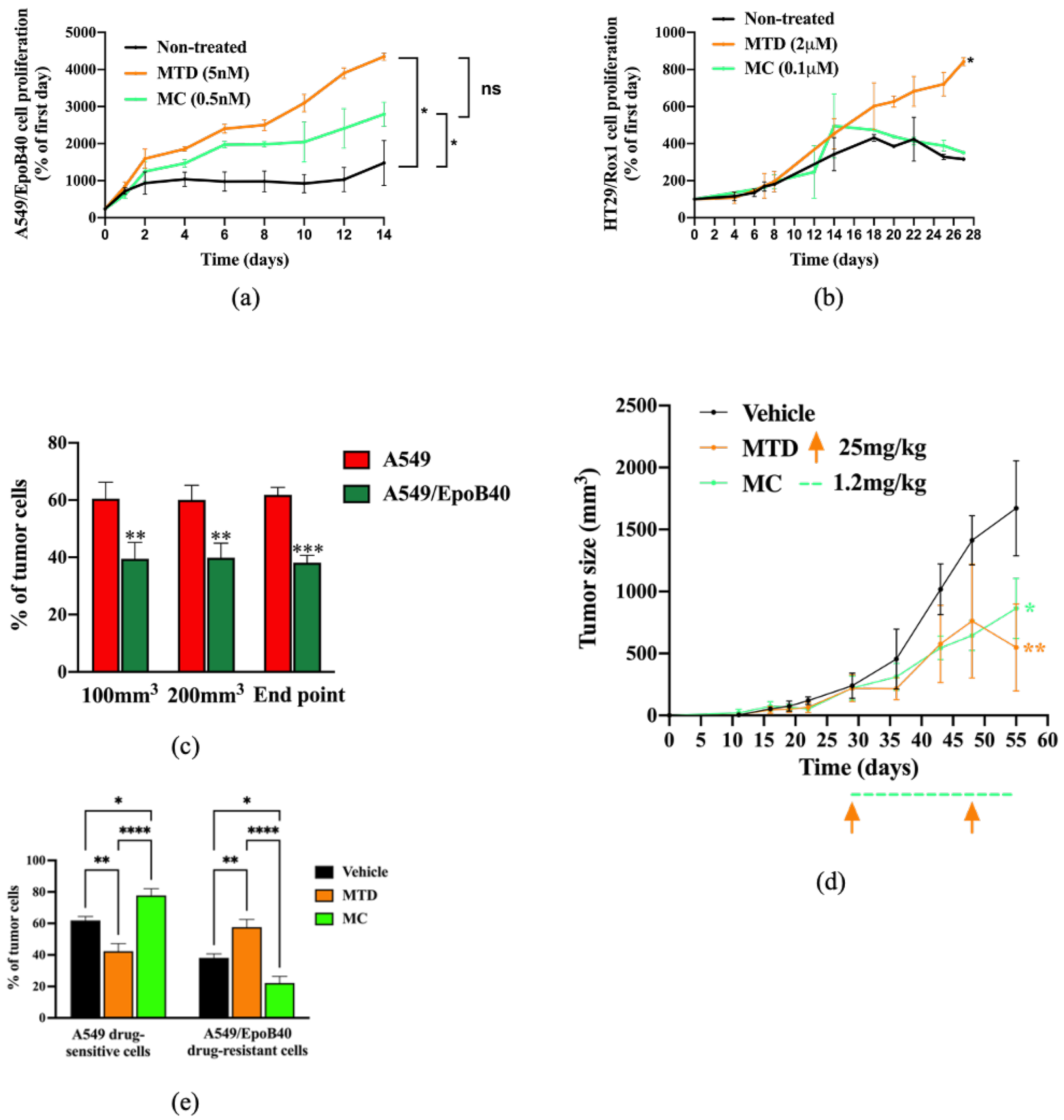
2.3. In Vitro and In Vivo Biological Validations of Mathematical Model Predictions
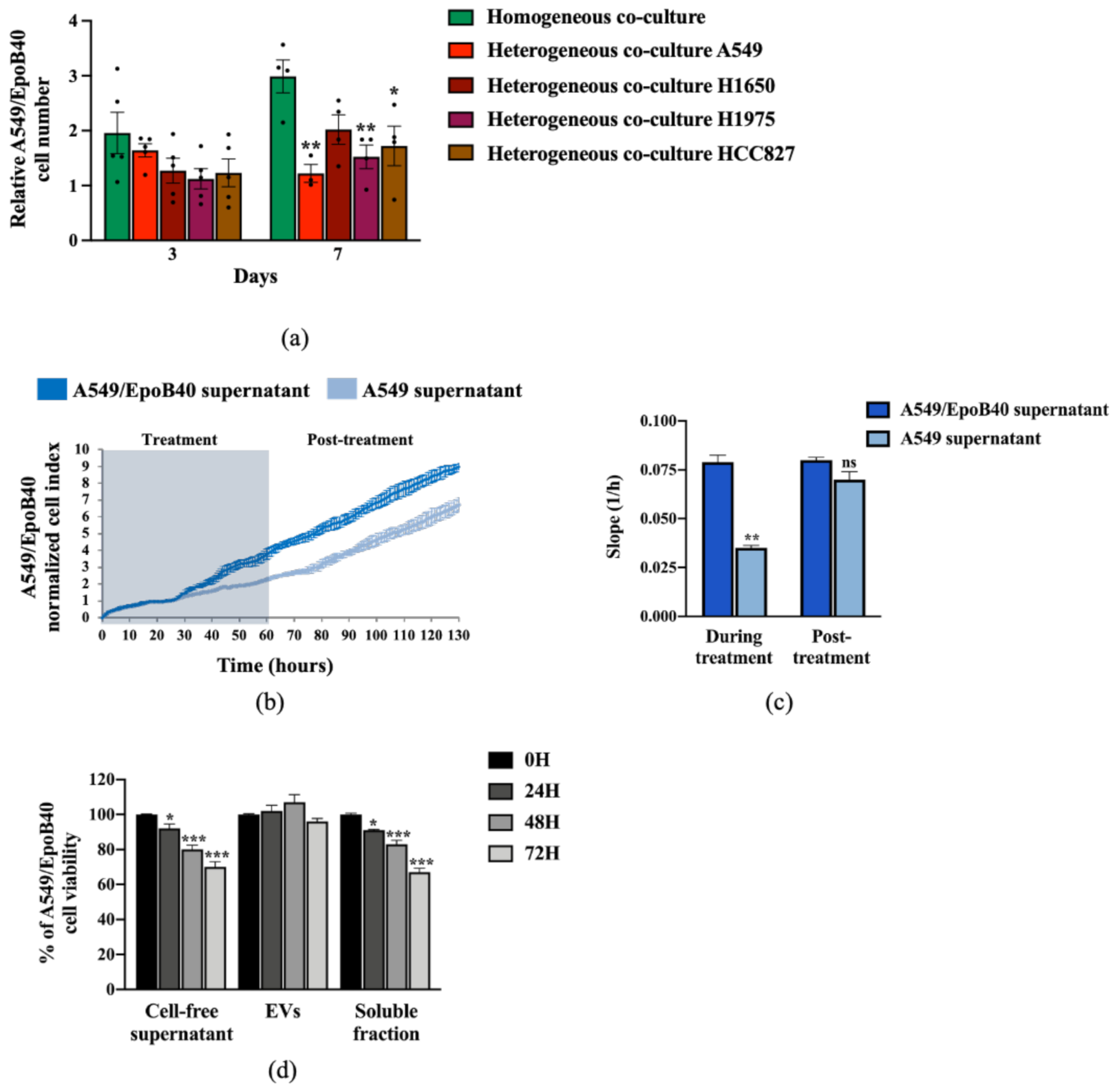
2.4. Drug-Sensitive Clones Control the Proliferation of Drug-Resistant Clones Through Indirect Cell-Cell Interaction
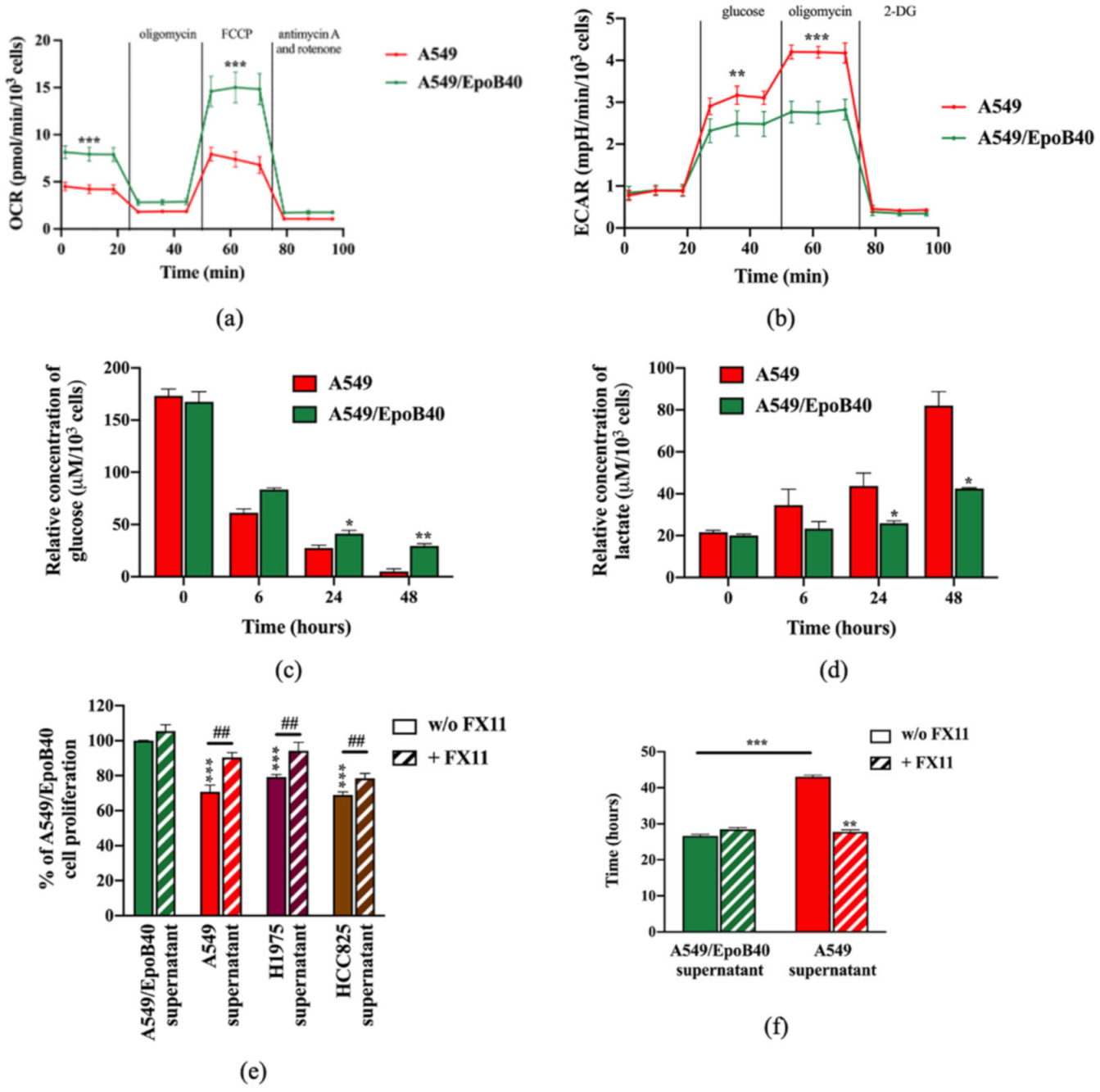
2.5. The Metabolic Activity of Drug-Sensitive Clones Is a Key Factor to Maintain Their Repression on Drug-Resistant Clones
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Drugs and Reagents
4.3. Establishment of Homo- and Heterogeneous 2D Co-Culture Models
4.4. Establishment of Homo- and Heterogeneous 3D Co-Culture Models
4.5. Cell Viability Assay
4.6. Proliferation Assay
4.7. Impedance Measurements
4.8. Transwell Co-Culture System
4.9. Extracellular Vesicles Isolation
4.10. Real-Time Metabolic Analysis
4.11. Glucose Consumption and Lactate Production Assay
4.12. In Vivo Studies
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A. A change of strategy in the war on cancer. Nature 2009, 459, 508–509. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriquez-Navas, P.M.; Wojtkowiak, J.W.; Gatenby, R.A. Application of evolutionary principles to cancer therapy. Cancer Res. 2015, 75, 4675–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Kavallaris, M.; André, N. Metronomic chemotherapy: New rationale for new directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef]
- Loven, D.; Hasnis, E.; Bertolini, F.; Shaked, Y. Low-dose metronomic chemotherapy: From past experience to new paradigms in the treatment of cancer. Drug Discov. Today 2013, 18, 193–201. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Grothey, A. Rationale for metronomic chemotherapy in phase III trials. Nat. Rev. Clin. Oncol. 2015, 12, 313–314. [Google Scholar] [CrossRef]
- Simsek, C.; Esin, E.; Yalcin, S. Metronomic chemotherapy: A systematic review of the literature and clinical experience. J. Oncol. 2019, 2019, 5483791. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-L.; Chang, M.-C.; Cheng, W.-F. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017, 400, 282–292. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Carrère, C. Optimization of an in vitro chemotherapy to avoid resistant tumours. J. Theor. Biol. 2017, 413, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, F.; Sahai, E. Cell communication networks in cancer invasion. Curr. Opin. Cell Biol. 2011, 23, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Brinton, L.T.; Sloane, H.S.; Kester, M.; Kelly, K.A. Formation and role of exosomes in cancer. Cell. Mol. Life Sci. 2015, 72, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Mishra, D.; Banerjee, D. Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Ma, S.; Xue, Y.; Hou, J.; Zhang, Y. LDH-A promotes malignant progression via activation of epithelial-to-mesenchymal transition and conferring stemness in muscle-invasive bladder cancer. Biochem. Biophys. Res. Commun. 2016, 469, 985–992. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Hirotsu, Y.; Amemiya, K.; Mochizuki, H.; Omata, M. Understanding intratumor heterogeneity and evolution in NSCLC and potential new therapeutic approach. Cancers 2018, 10, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quoix, E.; Zalcman, G.; Oster, J.-P.; Westeel, V.; Pichon, E.; Lavolé, A.; Dauba, J.; Debieuvre, D.; Souquet, P.J.; Bigay-Game, L.; et al. Carboplatin and weekly paclitaxel doublet chemotherapy compared with monotherapy in elderly patients with advanced non-small-cell lung cancer: IFCT-0501 randomised, phase 3 trial. Lancet 2011, 378, 1079–1088. [Google Scholar] [CrossRef]
- Socinski, M.A.; Bondarenko, I.; Karaseva, N.A.; Makhson, A.M.; Vynnychenko, I.; Okamoto, I.; Hon, J.K.; Hirsh, V.; Bhar, P.; Zhang, H.; et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: Final results of a phase III trial. J. Clin. Oncol. 2012, 30, 2055–2062. [Google Scholar] [CrossRef] [Green Version]
- D’Ascanio, M.; Pezzuto, A.; Fiorentino, C.; Sposato, B.; Bruno, P.; Grieco, A.; Mancini, R.; Ricci, A. Metronomic chemotherapy with vinorelbine produces clinical benefit and low toxicity in frail elderly patients affected by advanced non-small cell lung cancer. BioMed Res. Int. 2018, 2018, 6278403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortot, A.B.; Audigier-Valette, C.; Molinier, O.; Le Moulec, S.; Barlesi, F.; Zalcman, G.; Dumont, P.; Pouessel, D.; Poulet, C.; Fontaine-Delaruelle, C.; et al. Weekly paclitaxel plus bevacizumab versus docetaxel as second- or third-line treatment in advanced non-squamous non-small-cell lung cancer: Results of the IFCT-1103 ULTIMATE study. Eur. J. Cancer 1990, 131, 27–36. [Google Scholar] [CrossRef]
- Shu, Y.; Weng, S.; Zheng, S. Metronomic chemotherapy in non-small cell lung cancer (Review). Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- André, N.; Carré, M.; Pasquier, E. Metronomics: Towards personalized chemotherapy? Nat. Rev. Clin. Oncol. 2014, 11, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Cao, Y.; Lei, Z.; Yang, Z.; Zhang, B.; Huang, B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010, 70, 4850–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Orlandi, P.; di Desidero, T.; Salvia, G.; Muscatello, B.; Francia, G.; Bocci, G. Metronomic vinorelbine is directly active on non small cell lung cancer cells and sensitizes the EGFRL858R/T790M cells to reversible EGFR tyrosine kinase inhibitors. Biochem. Pharmacol. 2018, 152, 327–337. [Google Scholar] [CrossRef]
- Vives, M.; Ginestà, M.M.; Gracova, K.; Graupera, M.; Casanovas, O.; Capellà, G.; Serrano, T.; Laquente, B.; Vinals, F. Metronomic chemotherapy following the maximum tolerated dose is an effective anti-tumour therapy affecting angiogenesis, tumour dissemination and cancer stem cells. Int. J. Cancer 2013, 133, 2464–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkins, C.; Man, S.; Xu, P.; Shaked, Y.; Hicklin, D.J.; Kerbel, R.S. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007, 67, 3560–3564. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; de Jesús, K.; Mailankody, S. The high price of anticancer drugs: Origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef]
- Jardim, D.L.; de Melo, D.G.; Giles, F.J.; Kurzrock, R. Analysis of drug development paradigms for immune checkpoint inhibitors. Clin. Cancer Res. 2018, 24, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Rockne, R.C.; Hawkins-Daarud, A.; Swanson, K.R.; Sluka, J.P.; Glazier, J.A.; Macklin, P.; Hormuth, D.A., II; Jarrett, A.M.; Lima, E.A.B.F.; Oden, J.T.; et al. The 2019 mathematical oncology roadmap. Phys. Biol. 2019, 16, 041005. [Google Scholar] [CrossRef]
- Benzekry, S. Artificial intelligence and mechanistic modeling for clinical decision making in oncology. Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Barbolosi, D.; Meille, C.; Lombard, A.; Serdjebi, C.; Giacometti, S.; Padovani, L.; Pasquier, E.; Andre, N. Pharmacokinetics and pharmacodynamics-based mathematical modeling identifies an optimal protocol for metronomic chemotherapy. Cancer Res. 2017, 77, 4723–4733. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Imbs, D.-C.; Tomasini, P.; Greillier, L.; Galloux, M.; Testot-Ferry, A.; Garcia, M.; Elharrar, X.; Pelletier, A.; Andre, N.; et al. Mathematical modeling for phase I cancer trials: A study of metronomic vinorelbine for advanced non-small cell lung cancer (NSCLC) and mesothelioma patients. Oncotarget 2017, 8, 47161–47166. [Google Scholar] [CrossRef] [Green Version]
- West, J.B.; Dinh, M.N.; Brown, J.S.; Zhang, J.; Anderson, A.R.; Gatenby, R.A. Multidrug cancer therapy in metastatic castrate-resistant prostate cancer: An evolution-based strategy. Clin. Cancer Res. 2019, 25, 4413–4421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L.; et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef] [Green Version]
- Stanková, K.; Brown, J.S.; Dalton, W.S.; Gatenby, R.A. Optimizing cancer treatment using game theory: A review. JAMA Oncol. 2019, 5, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-Y.; Loo, T.Y.; Shen, J.-G.; Wang, N.; Wang, D.-M.; Yang, D.-P.; Mo, S.-L.; Guan, X.-Y.; Chen, J.-P. LDH-A silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res. Treat. 2012, 131, 791–800. [Google Scholar] [CrossRef]
- Savry, A.; Carre, M.; Berges, R.; Rovini, A.; Pobel, I.; Chacon, C.; Braguer, D.; Bourgarel-Rey, V. Bcl-2-enhanced efficacy of microtubule-targeting chemotherapy through BIM overexpression: Implications for cancer treatment. Neoplasia 2013, 15, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Grand, M.; Berges, R.; Pasquier, E.; Montero, M.-P.; Borge, L.; Carrier, A.; Vasseur, S.; Bourgarel, V.; Buric, D.; Andre, N.; et al. Akt targeting as a strategy to boost chemotherapy efficacy in non-small cell lung cancer through metabolism suppression. Sci. Rep. 2017, 7, 45136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timaner, M.; Beyar-Katz, O.; Shaked, Y. Analysis of the stromal cellular components of the solid tumor microenvironment using flow cytometry. Curr. Protoc. Cell Biol. 2016, 70, 19.18.1–19.18.12. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondarenko, M.; Le Grand, M.; Shaked, Y.; Raviv, Z.; Chapuisat, G.; Carrère, C.; Montero, M.-P.; Rossi, M.; Pasquier, E.; Carré, M.; et al. Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer. Cancers 2021, 13, 2239. https://doi.org/10.3390/cancers13092239
Bondarenko M, Le Grand M, Shaked Y, Raviv Z, Chapuisat G, Carrère C, Montero M-P, Rossi M, Pasquier E, Carré M, et al. Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer. Cancers. 2021; 13(9):2239. https://doi.org/10.3390/cancers13092239
Chicago/Turabian StyleBondarenko, Maryna, Marion Le Grand, Yuval Shaked, Ziv Raviv, Guillemette Chapuisat, Cécile Carrère, Marie-Pierre Montero, Mailys Rossi, Eddy Pasquier, Manon Carré, and et al. 2021. "Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer" Cancers 13, no. 9: 2239. https://doi.org/10.3390/cancers13092239
APA StyleBondarenko, M., Le Grand, M., Shaked, Y., Raviv, Z., Chapuisat, G., Carrère, C., Montero, M. -P., Rossi, M., Pasquier, E., Carré, M., & André, N. (2021). Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer. Cancers, 13(9), 2239. https://doi.org/10.3390/cancers13092239