
Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations

 , , , , ,
, , , , ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Sensitivity to Drugs
2.2. Loss of Function Studies
2.3. In Silico Analysis
2.4. Western Blot Analysis
2.5. Apoptosis Analysis
2.6. RNA Extraction and qRT-PCR Analysis
2.7. Statistical Analysis
3. Results
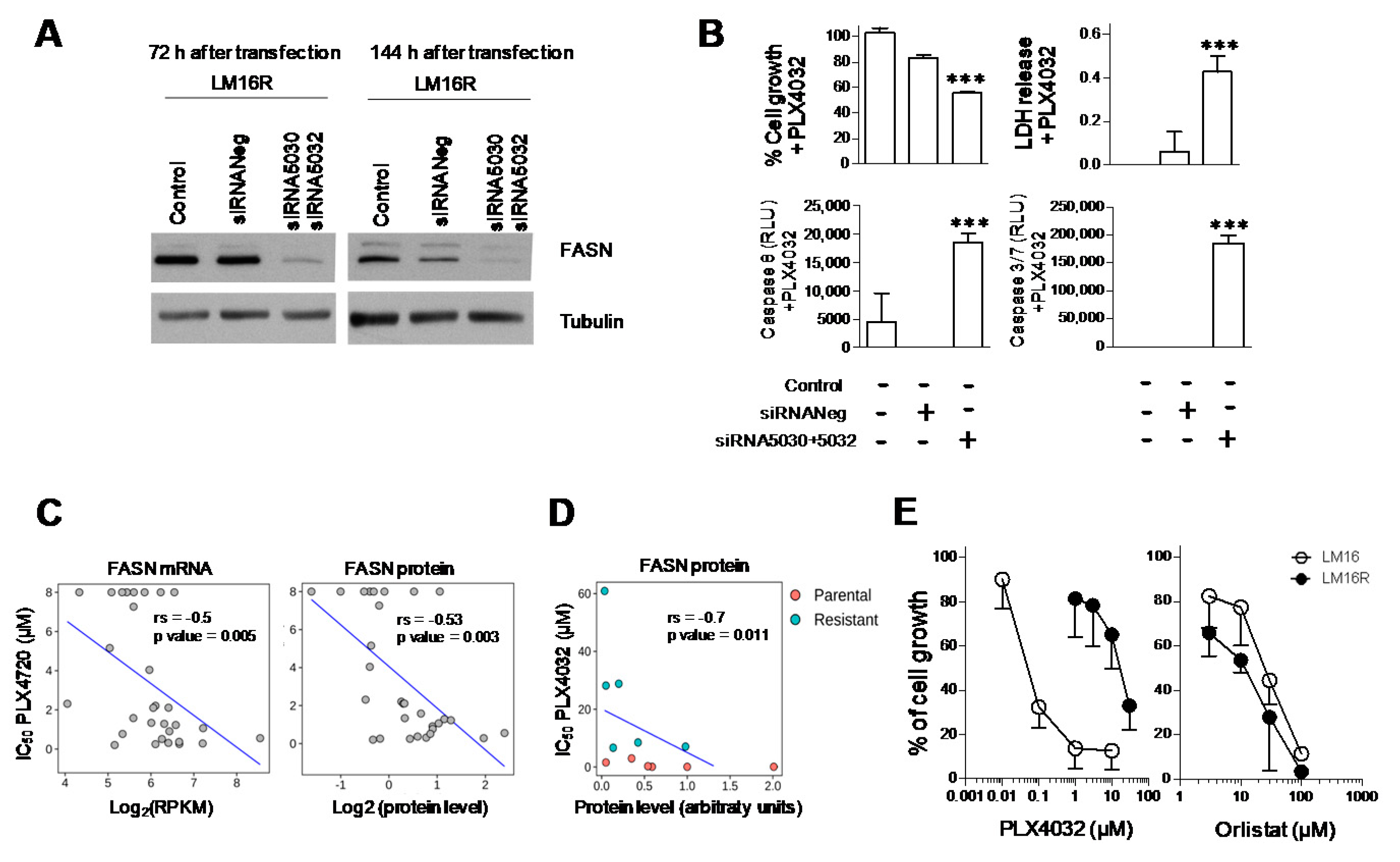
3.1. Knockdown of FASN in BRAFi-Resistant Melanoma Cells
3.2. Association between Dysregulation of FASN and Resistance to BRAF Inhibitors
3.3. Phenotype of LM16 R Cells Associates to Sensitivity to Orlistat
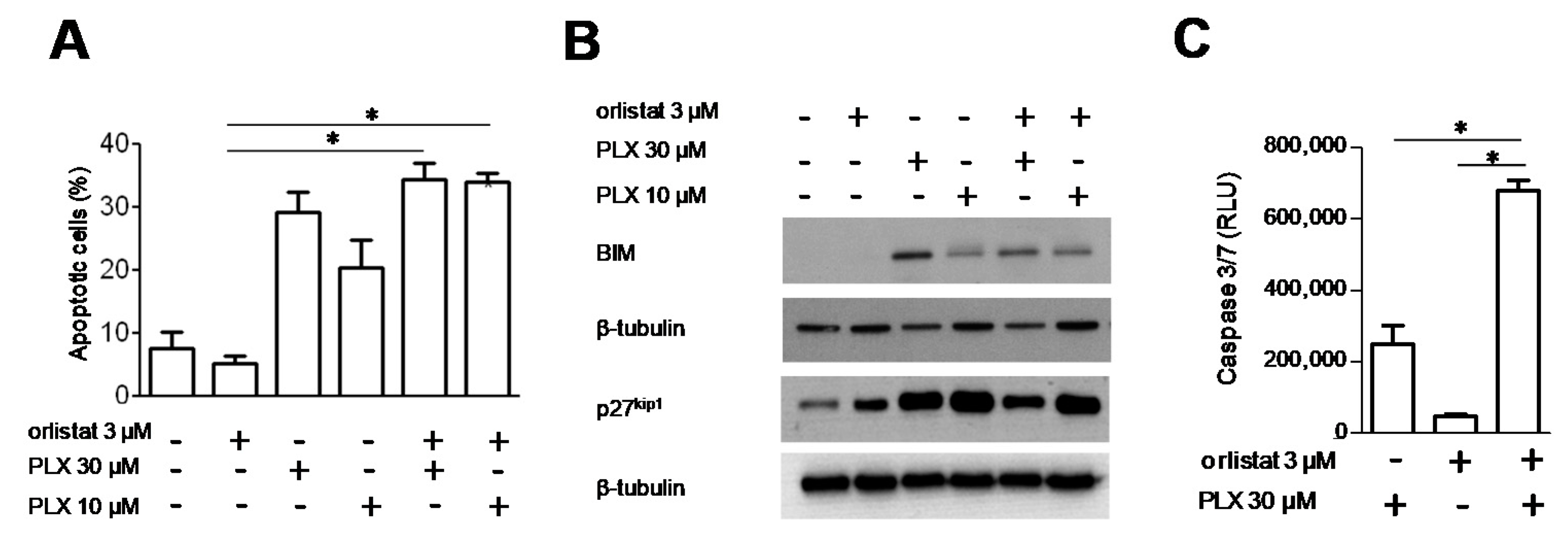
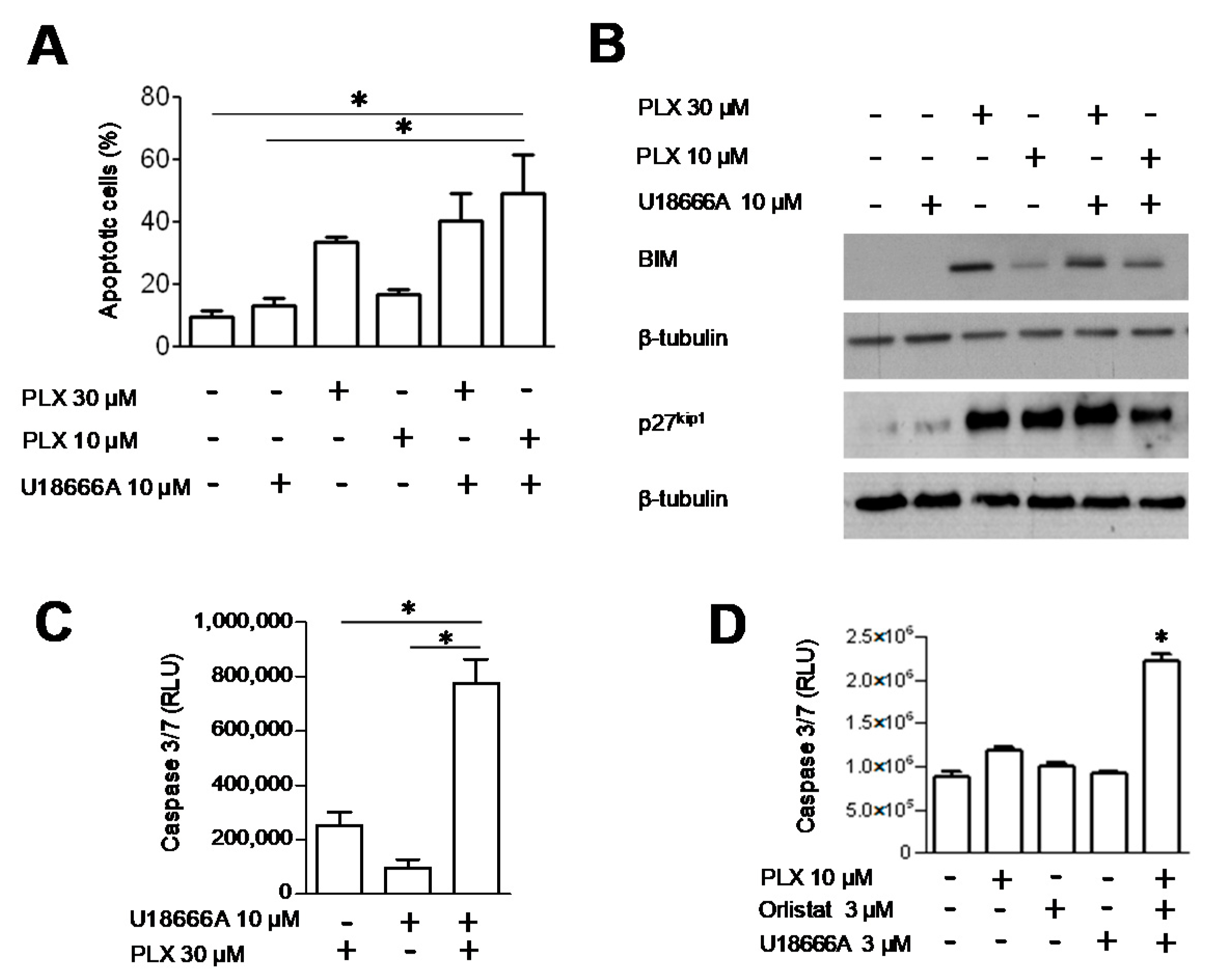
3.4. Analysis of Cell Response to the Combination of PLX4032 and Orlistat
3.5. DHCR24 Modulation upon FASN Inhibition and the Effect of DHCR24 Targeting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GLOBOCAN 2012. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Available online: http://globocan.iarc.fr (accessed on 1 April 2020).
- Gershenwald, J.E.; Scolyer, R.A. Melanoma Staging: American Joint Committee on Cancer (AJCC) 8th Edition and Beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nat. Cell Biol. 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta (BBA) Bioenerg. 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Innocenzi, D.; Alò, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty acid synthase expression in melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef]
- Kapur, P.; Rakheja, D.; Roy, L.C.; Hoang, M.P. Fatty acid synthase expression in cutaneous melanocytic neoplasms. Mod. Pathol. 2005, 18, 1107–1112. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Zhang, J.-T. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol. Cancer Ther. 2008, 7, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Eynde, K.V.D.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Garandeau, D.; Noujarède, J.; Leclerc, J.; Imbert, C.; Garcia, V.; Bats, M.-L.; Rambow, F.; Gilhodes, J.; Filleron, T.; Meyer, N.; et al. Targeting the Sphingosine 1-Phosphate Axis Exerts Potent Antitumor Activity in BRAFi-Resistant Melanomas. Mol. Cancer Ther. 2019, 18, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Miloslavskaya, I.; Demontis, S.; Maestro, R.; Galaktionov, K. Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nat. Cell Biol. 2004, 432, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehnle, K.; Crameri, A.; Kälin, R.E.; Luciani, P.; Benvenuti, S.; Peri, A.; Ratti, F.; Rodolfo, M.; Kulic, L.; Heppner, F.L.; et al. Prosurvival Effect of DHCR24/Seladin-1 in Acute and Chronic Responses to Oxidative Stress. Mol. Cell. Biol. 2007, 28, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greeve, I.; Hermans-Borgmeyer, I.; Brellinger, C.; Kasper, D.; Gomez-Isla, T.; Behl, C.; Levkau, B.; Nitsch, R.M. The Human DIMINUTO/DWARF1 Homolog Seladin-1 Confers Resistance to Alzheimer’s Disease-Associated Neurodegeneration and Oxidative Stress. J. Neurosci. 2000, 20, 7345–7352. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, D.; Vallacchi, V.; Campi, V.; Ranzani, T.; Daniotti, M.; Chiodini, E.; Fiorentini, S.; Greeve, I.; Prinetti, A.; Rivoltini, L.; et al. DHCR24 gene expression is upregulated in melanoma metastases and associated to resistance to oxidative stress-induced apoptosis. Int. J. Cancer 2005, 115, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, L.; Liu, R.; Song, Y.; Zhang, X.; Niu, W.; Kumar, A.K.; Guo, Z.; Hu, Z. Obesity-induced overexpression of miRNA-24 regulates cholesterol uptake and lipid metabolism by targeting SR-B1. Gene 2018, 668, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M.G.; Wang, J.; et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2019, 69, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Ballinger, A.; Peikin, S.R. Orlistat: Its current status as an anti-obesity drug. Eur. J. Pharmacol. 2002, 440, 109–117. [Google Scholar] [CrossRef]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schcolnik-Cabrera, A.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Taja-Chayeb, L.; Morales-Barcenas, R.; Trejo-Becerril, C.; Perez-Cardenas, E.; Gonzalez-Fierro, A.; Dueñas-González, A. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin. Investig. Drugs 2018, 27, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Daniotti, M.; Oggionni, M.; Ranzani, T.; Vallacchi, V.; Campi, V.; Di Stasi, D.; Della Torre, G.; Perrone, F.; Luoni, C.; Suardi, S.; et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene 2004, 23, 5968–5977. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Vallacchi, V.; Frigerio, S.; Deho, P.; Mondellini, P.; Perego, P.; Cassinelli, G.; Lanzi, C.; Testi, M.A.; Rivoltini, L.; et al. Identification of MET and SRC Activation in Melanoma Cell Lines Showing Primary Resistance to PLX4032. Neoplasia 2011, 13, 1132-IN17. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Corno, C.; Gatti, L.; Arrighetti, N.; Carenini, N.; Zaffaroni, N.; Lanzi, C.; Perego, P. Axl molecular targeting counteracts aggressiveness but not platinum-resistance of ovarian carcinoma cells. Biochem. Pharmacol. 2017, 136, 40–50. [Google Scholar] [CrossRef]
- Quan, X.; Chen, X.; Sun, D.; Xu, B.; Zhao, L.; Shi, X.; Liu, H.; Gao, B.; Lu, X. The mechanism of the effect of U18666a on blocking the activity of 3β-hydroxysterol Δ-24-reductase (DHCR24): Molecular dynamics simulation study and free energy analysis. J. Mol. Model. 2016, 22, 1–12. [Google Scholar] [CrossRef]
- Kusakabe, T.; Maeda, M.; Hoshi, N.; Sugino, T.; Watanabe, K.; Fukuda, T.; Suzuki, T. Fatty Acid Synthase Is Expressed Mainly in Adult Hormone-sensitive Cells or Cells with High Lipid Metabolism and in Proliferating Fetal Cells1. J. Histochem. Cytochem. 2000, 48, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.C.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.; Loda, M.; et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, Y.; Wang, W.; Chen, S.; Liu, T.; Jia, D.; Quan, X.; Sun, D.; Chang, A.K.; Gao, B. 3 β-Hydroxysteroid-Δ 24 Reductase (DHCR24) Protects Neuronal Cells from Apoptotic Cell Death Induced by Endoplasmic Reticulum (ER) Stress. PLoS ONE 2014, 9, e86753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Lai, F.; Thorne, R.F.; Yang, F.; Liu, H.; Hersey, P.; Zhang, X.D. MEK-Independent Survival of B-RAFV600E Melanoma Cells Selected for Resistance to Apoptosis Induced by the RAF Inhibitor PLX4720. Clin. Cancer Res. 2011, 17, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorenko, I.V.; Paraiso, K.H.; Smalley, K.S. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2015, 7, 4428–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CI ² | |
|---|---|---|
| PLX4032 | 3 µM orlistat | 10 µM orlistat |
| 30 µM | 0.22 ± 0.16 | 0.39 ± 0.11 |
| 10 µM | 0.68 ± 0.07 | 1.11 ± 0.35 |
| 3 µM | 1.39 ± 0.3 | 1.40 ± 0.53 |
| 1 µM | 4.3 ± 3.24 | 1.79 ± 0.62 |
| Variable | CI ² | |
|---|---|---|
| PLX4032 | 3 µM U18666A | 10 µM U18666A |
| 30 µM | 0.38 ± 0.15 | 0.30 ± 0.05 |
| 10 µM | 0.44 ± 0.10 | 0.41 ± 0.04 |
| 3 µM | 0.57 ± 0.18 | 0.67 ± 0.06 |
| 1 µM | 0.55 ± 0.02 | 0.79 ± 0.04 |
| PLX4032 | Orlistat (µM) | U18666A (µM) | CI 2 |
|---|---|---|---|
| 10 µM | 10 µM | 10 µM | 0.28 ± 0.17 |
| 10 µM | 10 µM | 3 µM | 0.37 ± 0.36 |
| 10 µM | 3 µM | 10 µM | 0.30 ± 0.26 |
| 10 µM | 3 µM | 3 µM | 0.31 ± 0.05 |
| 3 µM | 10 µM | 10 µM | 0.43 ± 0.00 |
| 3 µM | 10 µM | 3 µM | 0.64 ± 0.12 |
| 3 µM | 3 µM | 10 µM | 0.26 ± 0.25 |
| 3 µM | 3 µM | 3 µM | 0.61 ± 0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers 2021, 13, 2284. https://doi.org/10.3390/cancers13092284
Stamatakos S, Beretta GL, Vergani E, Dugo M, Corno C, Corna E, Tinelli S, Frigerio S, Ciusani E, Rodolfo M, et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers. 2021; 13(9):2284. https://doi.org/10.3390/cancers13092284
Chicago/Turabian StyleStamatakos, Serena, Giovanni Luca Beretta, Elisabetta Vergani, Matteo Dugo, Cristina Corno, Elisabetta Corna, Stella Tinelli, Simona Frigerio, Emilio Ciusani, Monica Rodolfo, and et al. 2021. "Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations" Cancers 13, no. 9: 2284. https://doi.org/10.3390/cancers13092284
APA StyleStamatakos, S., Beretta, G. L., Vergani, E., Dugo, M., Corno, C., Corna, E., Tinelli, S., Frigerio, S., Ciusani, E., Rodolfo, M., Perego, P., & Gatti, L. (2021). Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers, 13(9), 2284. https://doi.org/10.3390/cancers13092284