
Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment

 , , ,
, , , 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Prognostic Factors
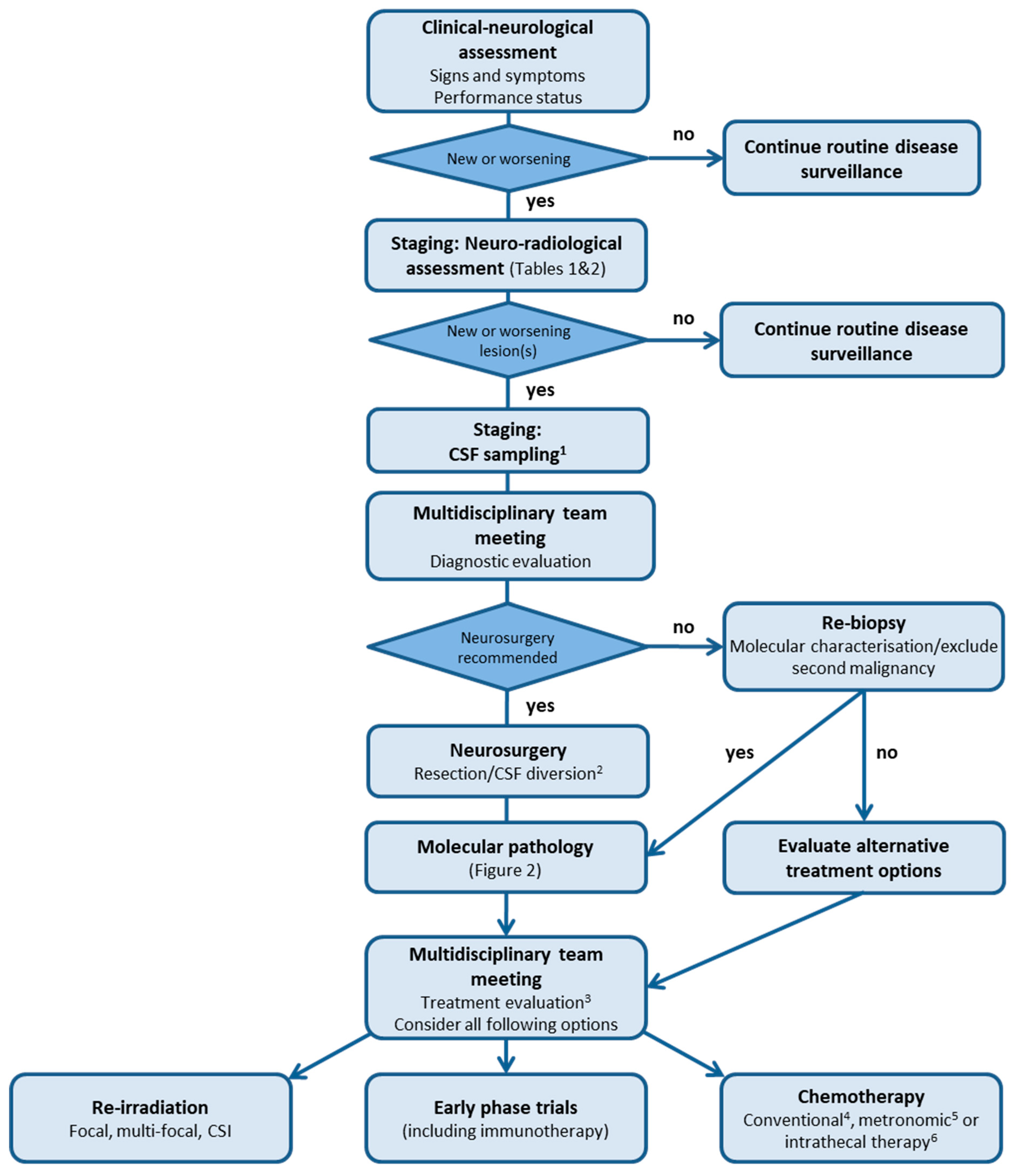
3. Clinical Diagnostic Process
3.1. Signs and Symptoms
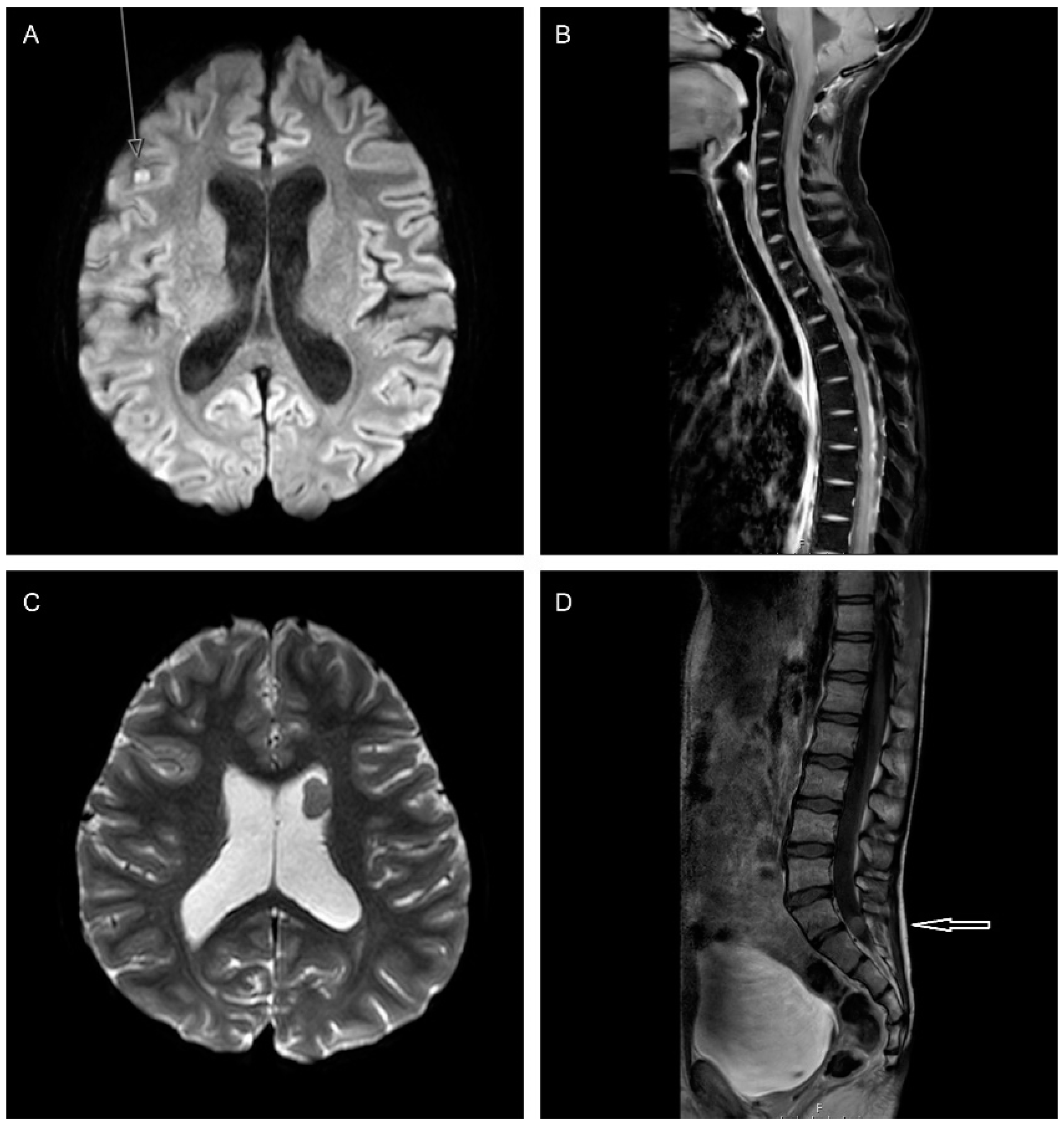
3.2. Staging: Neuro-Radiological Assessment
Response Assessment following Relapse
3.3. Staging: CSF Sampling
3.4. Molecular Pathology
4. Intention of Treatment
5. Treatment Modalities and Considerations
5.1. Neurosurgery
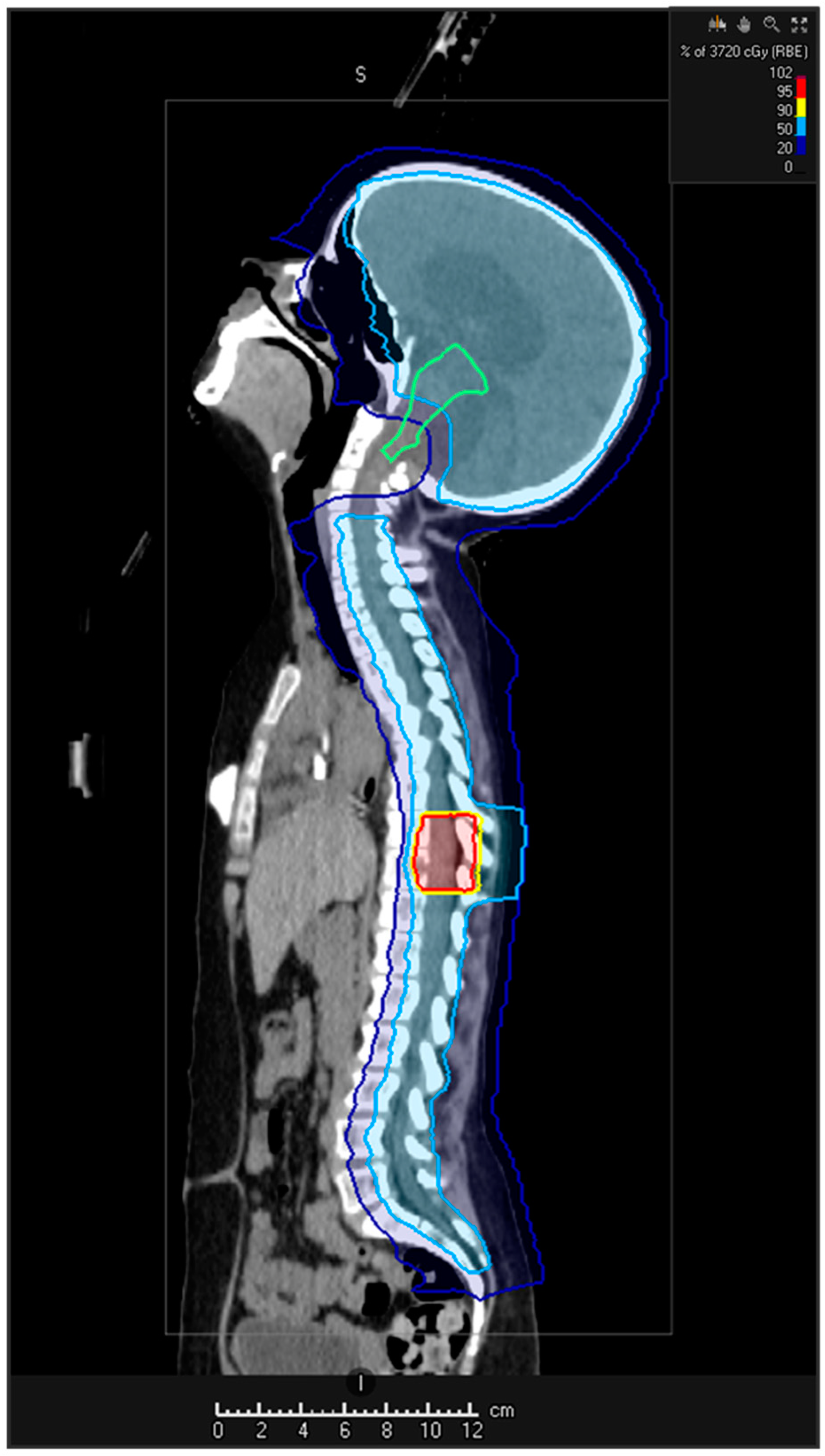
5.2. Re-Irradiation
5.2.1. Re-Irradiation Doses and Volume
5.2.2. Techniques
5.2.3. Stereotactic Radiotherapy/Radiosurgery
5.2.4. Intensity-Modulated Radiation Therapy (IMRT)
5.2.5. Proton Beam Therapy
5.3. Chemotherapy
5.3.1. High-Dose Chemotherapy
5.3.2. Conventional Chemotherapy
5.3.3. Temozolomide, Irinotecan and Bevacizumab
5.3.4. Temozolomide and Irinotecan (TEMIRI)
5.3.5. Temozolomide and Topotecan (TOTEM)
5.3.6. Temozolomide
5.3.7. Metronomic Chemotherapy
5.3.8. Etoposide
5.3.9. Metronomic and Targeted Antiangiogenesis Therapy for Children with Recurrent/Progressive Medulloblastoma (MEMMAT)
5.3.10. Modified MEMMAT
5.3.11. Combined Oral Metronomic Biodifferentiating Antiangiogenic Treatment (COMBAT)
5.3.12. Temozolomide and Etoposide
5.4. Targeted Therapies
5.4.1. MBSHH: Smoothened Inhibitors
5.4.2. MBGroup3- and MBGroup4-Targeted Therapies
5.5. Immunotherapy
5.5.1. Checkpoint Inhibitors
5.5.2. Oncolytic Viruses
5.5.3. Chimeric Antigen Receptor Therapy (CAR T-Cell Therapy)
5.6. Intrathecal Therapies
5.6.1. Intrathecal Chemotherapy
5.6.2. Intrathecal Immunotherapy
6. Supportive Care and Follow-Up Investigations
7. Future Developments
7.1. Drug–Target Matched Clinical Trials
7.2. Liquid Biopsies
7.3. Modelling Strategies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84. [Google Scholar] [CrossRef]
- Hill, R.M.; Richardson, S.; Schwalbe, E.C.; Hicks, D.; Lindsey, J.C.; Crosier, S.; Rafiee, G.; Grabovska, Y.; Wharton, S.B.; Jacques, T.S.; et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: A multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 865–874. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Sabel, M.; Fleischhack, G.; Tippelt, S.; Gustafsson, G.; Doz, F.; Kortmann, R.; Massimino, M.; Navajas, A.; von Hoff, K.; Rutkowski, S.; et al. Relapse patterns and outcome after relapse in standard risk medulloblastoma: A report from the HIT-SIOP-PNET4 study. J. Neuro-Oncol. 2016, 129, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Pizer, B.; Donachie, P.H.; Robinson, K.; Taylor, R.E.; Michalski, A.; Punt, J.; Ellison, D.W.; Picton, S. Treatment of recurrent central nervous system primitive neuroectodermal tumours in children and adolescents: Results of a Children’s Cancer and Leukaemia Group study. Eur. J. Cancer 2011, 47, 1389–1397. [Google Scholar] [CrossRef]
- Richardson, S.; Hill, R.M.; Kui, C.; Lindsey, J.C.; Grabovksa, Y.; Keeling, C.; Pease, L.; Bashton, M.; Crosier, S.; Vinci, M.; et al. Emergence and maintenance of actionable genetic drivers at medulloblastoma relapse. Neuro-Oncology 2021, noab178. [Google Scholar] [CrossRef]
- Le Teuff, G.; Castaneda-Heredia, A.; Dufour, C.; Jaspan, T.; Calmon, R.; Devos, A.; McHugh, K.; Leblond, P.; Frappaz, D.; Aerts, I.; et al. Phase II study of temozolomide and topotecan (TOTEM) in children with relapsed or refractory extracranial and central nervous system tumors including medulloblastoma with post hoc Bayesian analysis: A European ITCC study. Pediatr. Blood Cancer 2020, 67, e28032. [Google Scholar] [CrossRef]
- Bode, U.; Zimmermann, M.; Moser, O.; Rutkowski, S.; Warmuth-Metz, M.; Pietsch, T.; Kortmann, R.D.; Faldum, A.; Fleischhack, G. Treatment of recurrent primitive neuroectodermal tumors (PNET) in children and adolescents with high-dose chemotherapy (HDC) and stem cell support: Results of the HITREZ 97 multicentre trial. J. Neuro-Oncol. 2014, 120, 635–642. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.; Schwalbe, E.C.; Williamson, D.; Sill, M.; Hovestadt, V.; Mynarek, M.; Rutkowski, S.; Robinson, G.W.; Gajjar, A.; Cavalli, F.; et al. Second-generation molecular subgrouping of medulloblastoma: An international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019, 138, 309–326. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Kumar, R.; Smith, K.S.; Deng, M.; Terhune, C.; Robinson, G.W.; Orr, B.A.; Liu, A.P.Y.; Lin, T.; Billups, C.A.; Chintagumpala, M.; et al. Clinical Outcomes and Patient-Matched Molecular Composition of Relapsed Medulloblastoma. J. Clin. Oncol. 2021, 39, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Vo, K.T.; Parsons, D.W.; Seibel, N.L. Precision Medicine in Pediatric Oncology. Surg. Oncol. Clin. N. Am. 2020, 29, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.J.; Geoerger, B.; Janeway, K.A. Precision medicine in pediatric oncology. Curr. Opin. Pediatr. 2018, 30, 17–24. [Google Scholar] [CrossRef]
- George, S.L.; Izquierdo, E.; Campbell, J.; Koutroumanidou, E.; Proszek, P.; Jamal, S.; Hughes, D.; Yuan, L.; Marshall, L.V.; Carceller, F.; et al. A tailored molecular profiling programme for children with cancer to identify clinically actionable genetic alterations. Eur. J. Cancer 2019, 121, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Janeway, K.A.; Patton, D.; Coffey, B.; Williams, P.M.; Hamilton, S.R.; Purkayastha, A.; Tsongalis, G.J.; Routbort, M.; Gastier-Foster, J.M.; et al. Identification of targetable molecular alterations in the NCI-COG Pediatric MATCH trial. J. Clin. Oncol. 2019, 37 (Suppl. S15), 10011. [Google Scholar] [CrossRef]
- van Tilburg, C.M.; Pfaff, E.; Pajtler, K.W.; Langenberg, K.P.S.; Fiesel, P.; Jones, B.C.; Balasubramanian, G.P.; Stark, S.; Johann, P.D.; Blattner-Johnson, M.; et al. The Pediatric Precision Oncology INFORM Registry: Clinical Outcome and Benefit for Patients with Very High-Evidence Targets. Cancer Discov. 2021, 11, 2764–2779. [Google Scholar] [CrossRef]
- Deng, M.Y.; Sturm, D.; Pfaff, E.; Sill, M.; Stichel, D.; Balasubramanian, G.P.; Tippelt, S.; Kramm, C.; Donson, A.M.; Green, A.L.; et al. Radiation-induced gliomas represent H3-/IDH-wild type pediatric gliomas with recurrent PDGFRA amplification and loss of CDKN2A/B. Nat. Commun. 2021, 12, 5530. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huybrechts, S.; Le Teuff, G.; Tauziede-Espariat, A.; Rossoni, C.; Chivet, A.; Indersie, E.; Varlet, P.; Puget, S.; Abbas, R.; Ayrault, O.; et al. Prognostic Clinical and Biologic Features for Overall Survival after Relapse in Childhood Medulloblastoma. Cancers 2020, 13, 53. [Google Scholar] [CrossRef]
- Rutkowski, S.; Modena, P.; Williamson, D.; Kerl, K.; Nysom, K.; Pizer, B.; Bartels, U.; Puget, S.; Doz, F.; Michalski, A.; et al. Biological material collection to advance translational research and treatment of children with CNS tumours: Position paper from the SIOPE Brain Tumour Group. Lancet Oncol. 2018, 19, e419–e428. [Google Scholar] [CrossRef]
- Saunders, D.E.; Hayward, R.D.; Phipps, K.P.; Chong, W.K.; Wade, A.M. Surveillance neuroimaging of intracranial medulloblastoma in children: How effective, how often, and for how long? J. Neurosurg. 2003, 99, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtech, M.; Morris, C.G.; Indelicato, D.J.; Rutenberg, M.S.; Amdur, R.J. Patterns of Failure in Patients With Adult Medulloblastoma Presenting Without Extraneural Metastasis. Am. J. Clin. Oncol. 2018, 41, 1015–1018. [Google Scholar] [CrossRef]
- Avula, S.; Peet, A.; Morana, G.; Morgan, P.; Warmuth-Metz, M.; Jaspan, T.; European Society for Paediatric Oncology Brain Tumour Imaging Group. European Society for Paediatric Oncology (SIOPE) MRI guidelines for imaging patients with central nervous system tumours. Child’s Nerv. Syst. 2021, 37, 2497–2508. [Google Scholar] [CrossRef]
- Warren, K.E.; Vezina, G.; Poussaint, T.Y.; Warmuth-Metz, M.; Chamberlain, M.C.; Packer, R.J.; Brandes, A.A.; Reiss, M.; Goldman, S.; Fisher, M.J.; et al. Response assessment in medulloblastoma and leptomeningeal seeding tumors: Recommendations from the Response Assessment in Pediatric Neuro-Oncology committee. Neuro-Oncology 2018, 20, 13–23. [Google Scholar] [CrossRef]
- Kremer, S.; Abu Eid, M.; Bierry, G.; Bogorin, A.; Koob, M.; Dietemann, J.L.; Fruehlich, S. Accuracy of delayed post-contrast FLAIR MR imaging for the diagnosis of leptomeningeal infectious or tumoral diseases. J. Neuroradiol. 2006, 33, 285–291. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Coley, S.C.; Romanowski, C.A.; Hodgson, T.; Wilkinson, I.D. Contrast-enhanced fluid-attenuated inversion recovery imaging for leptomeningeal disease in children. AJNR Am. J. Neuroradiol. 2003, 24, 719–723. [Google Scholar]
- Kushnirsky, M.; Nguyen, V.; Katz, J.S.; Steinklein, J.; Rosen, L.; Warshall, C.; Schulder, M.; Knisely, J.P. Time-delayed contrast-enhanced MRI improves detection of brain metastases and apparent treatment volumes. J. Neurosurg. 2016, 124, 489–495. [Google Scholar] [CrossRef]
- Cohen-Inbar, O.; Xu, Z.; Dodson, B.; Rizvi, T.; Durst, C.R.; Mukherjee, S.; Sheehan, J.P. Time-delayed contrast-enhanced MRI improves detection of brain metastases: A prospective validation of diagnostic yield. J. Neuro-Oncol. 2016, 130, 485–494. [Google Scholar] [CrossRef]
- Buch, K.; Caruso, P.; Ebb, D.; Rincon, S. Balanced Steady-State Free Precession Sequence (CISS/FIESTA/3D Driven Equilibrium Radiofrequency Reset Pulse) Increases the Diagnostic Yield for Spinal Drop Metastases in Children with Brain Tumors. AJNR Am. J. Neuroradiol. 2018, 39, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, L.L.; Jones, R.A.; Palasis, S.; Aguilera, D.; Porter, D.A. Drop metastases to the pediatric spine revealed with diffusion-weighted MR imaging. Pediatr. Radiol. 2012, 42, 1009–1013. [Google Scholar] [CrossRef]
- Li, X.; Qu, J.R.; Luo, J.P.; Li, J.; Zhang, H.K.; Shao, N.N.; Kwok, K.; Zhang, S.N.; Li, Y.L.; Liu, C.C.; et al. Effect of intravenous gadolinium-DTPA on diffusion-weighted imaging of brain tumors: A short temporal interval assessment. J. Magn. Reson. Imaging 2014, 40, 616–621. [Google Scholar] [CrossRef]
- Chiaravalloti, A.; Filippi, L.; Ricci, M.; Cimini, A.; Schillaci, O. Molecular Imaging in Pediatric Brain Tumors. Cancers 2019, 11, 1853. [Google Scholar] [CrossRef] [Green Version]
- Cistaro, A.; Albano, D.; Alongi, P.; Laudicella, R.; Pizzuto, D.A.; Formica, G.; Romagnolo, C.; Stracuzzi, F.; Frantellizzi, V.; Piccardo, A.; et al. The Role of PET in Supratentorial and Infratentorial Pediatric Brain Tumors. Curr. Oncol. 2021, 28, 2481–2495. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [Green Version]
- Le Fevre, C.; Constans, J.M.; Chambrelant, I.; Antoni, D.; Bund, C.; Leroy-Freschini, B.; Schott, R.; Cebula, H.; Noel, G. Pseudoprogression versus true progression in glioblastoma patients: A multiapproach literature review. Part 2—Radiological features and metric markers. Crit. Rev. Oncol. Hematol. 2021, 159, 103230. [Google Scholar] [CrossRef]
- Sanders, R.P.; Onar, A.; Boyett, J.M.; Broniscer, A.; Morris, E.B.; Qaddoumi, I.; Armstrong, G.T.; Boop, F.A.; Sanford, R.A.; Kun, L.E.; et al. M1 Medulloblastoma: High risk at any age. J. Neuro-Oncol. 2008, 90, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Fouladi, M.; Gajjar, A.; Boyett, J.M.; Walter, A.W.; Thompson, S.J.; Merchant, T.E.; Jenkins, J.J.; Langston, J.W.; Liu, A.; Kun, L.E.; et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J. Clin. Oncol. 1999, 17, 3234–3237. [Google Scholar] [CrossRef]
- Straccia, P.; Fadda, G.; Pierconti, F. Comparison between cytospin and liquid-based cytology in cerebrospinal fluid diagnosis of neoplastic diseases: A single institution experience. Cytopathology 2019, 30, 236–240. [Google Scholar] [CrossRef]
- Damiani, D.; Suciu, V.; Andreiuolo, F.; Calderaro, J.; Vielh, P. Young investigator challenge: Cytomorphologic analysis of cerebrospinal fluid in 70 pediatric patients with medulloblastoma and review of the literature focusing on novel diagnostic and prognostic tests. Cancer Cytopathol. 2015, 123, 644–649. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Naran, S.; Lallu, S.; Fauck, R. Cytodiagnosis of neoplasms of the central nervous system in cerebrospinal fluid samples with an application of selective immunostains in differentiation. Cytopathology 2004, 15, 38–43. [Google Scholar] [CrossRef]
- Gajjar, A.; Fouladi, M.; Walter, A.W.; Thompson, S.J.; Reardon, D.A.; Merchant, T.E.; Jenkins, J.J.; Liu, A.; Boyett, J.M.; Kun, L.E.; et al. Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors. J. Clin. Oncol. 1999, 17, 1825–1828. [Google Scholar] [CrossRef]
- Chamberlain, M.C.; Kormanik, P.A.; Glantz, M.J. A comparison between ventricular and lumbar cerebrospinal fluid cytology in adult patients with leptomeningeal metastases. Neuro-Oncology 2001, 3, 42–45. [Google Scholar] [CrossRef]
- Tumani, H.; Petereit, H.F.; Gerritzen, A.; Gross, C.C.; Huss, A.; Isenmann, S.; Jesse, S.; Khalil, M.; Lewczuk, P.; Lewerenz, J.; et al. S1 guidelines “lumbar puncture and cerebrospinal fluid analysis” (abridged and translated version). Neurol. Res. Pract. 2020, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.G.; Kepner, J.L.; Goldthwaite, P.T.; Kun, L.E.; Duffner, P.K.; Friedman, H.S.; Strother, D.R.; Burger, P.C. Histopathologic grading of medulloblastomas: A Pediatric Oncology Group study. Cancer 2002, 94, 552–560. [Google Scholar] [CrossRef] [PubMed]
- McManamy, C.S.; Lamont, J.M.; Taylor, R.E.; Cole, M.; Pearson, A.D.; Clifford, S.C.; Ellison, D.W.; United Kingdom Children’s Cancer Study Group. Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J. Neuropathol. Exp. Neurol. 2003, 62, 627–632. [Google Scholar] [CrossRef] [Green Version]
- McManamy, C.S.; Pears, J.; Weston, C.L.; Hanzely, Z.; Ironside, J.W.; Taylor, R.E.; Grundy, R.G.; Clifford, S.C.; Ellison, D.W. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007, 17, 151–164. [Google Scholar] [CrossRef]
- Massimino, M.; Antonelli, M.; Gandola, L.; Miceli, R.; Pollo, B.; Biassoni, V.; Schiavello, E.; Buttarelli, F.R.; Spreafico, F.; Collini, P.; et al. Histological variants of medulloblastoma are the most powerful clinical prognostic indicators. Pediatr. Blood Cancer 2013, 60, 210–216. [Google Scholar] [CrossRef]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. World Health Organization Classification of Tumours of the Central Nervous System, Revised, 4th ed.; IARC: Lyon, France, 2016; Volume 1. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Liu, A.P.Y.; Northcott, P.A. Medulloblastoma genomics in the modern molecular era. Brain Pathol. 2020, 30, 679–690. [Google Scholar] [CrossRef]
- Hovestadt, V.; Ayrault, O.; Swartling, F.J.; Robinson, G.W.; Pfister, S.M.; Northcott, P.A. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 2020, 20, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef]
- Pietsch, T.; Haberler, C. Update on the integrated histopathological and genetic classification of medulloblastoma—A practical diagnostic guideline. Clin. Neuropathol. 2016, 35, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Korshunov, A.; Sahm, F.; Zheludkova, O.; Golanov, A.; Stichel, D.; Schrimpf, D.; Ryzhova, M.; Potapov, A.; Habel, A.; Meyer, J.; et al. DNA-methylation profiling is a method of choice for molecular verification of pediatric WNT activated medulloblastomas. Neuro-Oncology 2018, 21, 214–221. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Liu, A.P.Y.; Orr, B.A.; Northcott, P.A.; Robinson, G.W. Advances in the classification of pediatric brain tumors through DNA methylation profiling: From research tool to frontline diagnostic. Cancer 2018, 124, 4168–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waszak, S.M.; Robinson, G.W.; Gudenas, B.L.; Smith, K.S.; Forget, A.; Kojic, M.; Garcia-Lopez, J.; Hadley, J.; Hamilton, K.V.; Indersie, E.; et al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 2020, 580, 396–401. [Google Scholar] [CrossRef]
- Huq, A.J.; Walsh, M.; Rajagopalan, B.; Finlay, M.; Trainer, A.H.; Bonnet, F.; Sevenet, N.; Winship, I.M. Mutations in SUFU and PTCH1 genes may cause different cutaneous cancer predisposition syndromes: Similar, but not the same. Fam. Cancer 2018, 17, 601–606. [Google Scholar] [CrossRef]
- Orr, B.A.; Clay, M.R.; Pinto, E.M.; Kesserwan, C. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathol. 2020, 139, 669–687. [Google Scholar] [CrossRef]
- Kim, B.; Tabori, U.; Hawkins, C. An update on the CNS manifestations of brain tumor polyposis syndromes. Acta Neuropathol. 2020, 139, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Bourdeaut, F.; Miquel, C.; Richer, W.; Grill, J.; Zerah, M.; Grison, C.; Pierron, G.; Amiel, J.; Krucker, C.; Radvanyi, F.; et al. Rubinstein-Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr. Blood Cancer 2014, 61, 383–386. [Google Scholar] [CrossRef]
- Huang, J.; Grotzer, M.A.; Watanabe, T.; Hewer, E.; Pietsch, T.; Rutkowski, S.; Ohgaki, H. Mutations in the Nijmegen breakage syndrome gene in medulloblastomas. Clin. Cancer Res. 2008, 14, 4053–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugieres, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Begemann, M.; Waszak, S.M.; Robinson, G.W.; Jager, N.; Sharma, T.; Knopp, C.; Kraft, F.; Moser, O.; Mynarek, M.; Guerrini-Rousseau, L.; et al. Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J. Clin. Oncol. 2020, 38, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Baroni, L.V.; Freytes, C.; Fernandez Ponce, N.; Oller, A.; Pinto, N.; Gonzalez, A.; Maldonado, F.R.; Sampor, C.; Rugilo, C.; Lubieniecki, F.; et al. Craniospinal irradiation as part of re-irradiation for children with recurrent medulloblastoma. J. Neuro-Oncol. 2021, 155, 53–61. [Google Scholar] [CrossRef]
- Gits, H.C.; Anderson, M.; Stallard, S.; Pratt, D.; Zon, B.; Howell, C.; Kumar-Sinha, C.; Vats, P.; Kasaian, K.; Polan, D.; et al. Medulloblastoma therapy generates risk of a poorly-prognostic H3 wild-type subgroup of diffuse intrinsic pontine glioma: A report from the International DIPG Registry. Acta. Neuropathol. Commun. 2018, 6, 67. [Google Scholar] [CrossRef]
- Neglia, J.P.; Robison, L.L.; Stovall, M.; Liu, Y.; Packer, R.J.; Hammond, S.; Yasui, Y.; Kasper, C.E.; Mertens, A.C.; Donaldson, S.S.; et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2006, 98, 1528–1537. [Google Scholar] [CrossRef]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [CrossRef]
- Packer, R.J.; Zhou, T.; Holmes, E.; Vezina, G.; Gajjar, A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: Results of Children’s Oncology Group trial A9961. Neuro-Oncology 2013, 15, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Phi, J.H.; Park, A.K.; Lee, S.; Choi, S.A.; Baek, I.P.; Kim, P.; Kim, E.H.; Park, H.C.; Kim, B.C.; Bhak, J.; et al. Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. Acta Neuropathol. 2018, 135, 939–953. [Google Scholar] [CrossRef]
- Nantavithya, C.; Paulino, A.C.; Liao, K.; Woodhouse, K.D.; McGovern, S.L.; Grosshans, D.R.; McAleer, M.F.; Khatua, S.; Chintagumpala, M.M.; Majd, N.; et al. Observed-to-expected incidence ratios of second malignant neoplasms after radiation therapy for medulloblastoma: A Surveillance, Epidemiology, and End Results analysis. Cancer 2021, 127, 2368–2375. [Google Scholar] [CrossRef]
- Lopez, G.Y.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Clarke, J.; Oberheim Bush, N.A.; Taylor, J.; Chang, S.; et al. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol. 2019, 137, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Drezner, N.; Hardy, K.K.; Wells, E.; Vezina, G.; Ho, C.Y.; Packer, R.J.; Hwang, E.I. Treatment of pediatric cerebral radiation necrosis: A systematic review. J. Neuro-Oncol. 2016, 130, 141–148. [Google Scholar] [CrossRef]
- Loiacono, F.; Morra, A.; Venturini, S.; Balestreri, L. Abdominal metastases of medulloblastoma related to a ventriculoperitoneal shunt. AJR Am. J. Roentgenol. 2006, 186, 1548–1550. [Google Scholar] [CrossRef]
- Arias-Garzon, W.; Pacheco-Barsallo, F.; Trujillo-Jacome, C. Peritoneal metastases of medulloblastoma due to placement of ventriculoperitoneal shunt in an adult patient. Cir. Cir. 2011, 79, 458–463. [Google Scholar] [PubMed]
- Pettersson, D.; Schmitz, K.R.; Pollock, J.M.; Hopkins, K.L. Medulloblastoma: Seeding of VP shunt tract and peritoneum. Clin. Pract. 2012, 2, e37. [Google Scholar] [CrossRef]
- Wetmore, C.; Herington, D.; Lin, T.; Onar-Thomas, A.; Gajjar, A.; Merchant, T.E. Reirradiation of recurrent medulloblastoma: Does clinical benefit outweigh risk for toxicity? Cancer 2014, 120, 3731–3737. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.D.; Rashid, A.S.; Chen, Q.; Villar, R.C.; Kobyzeva, D.; Nilsson, K.; Dieckmann, K.; Nechesnyuk, A.; Ermoian, R.; Alcorn, S.; et al. Reirradiation for Recurrent Pediatric Central Nervous System Malignancies: A Multi-institutional Review. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 634–641. [Google Scholar] [CrossRef]
- Constine, L.S.; Olch, A.J.; Jackson, A.; Hua, C.H.; Ronckers, C.M.; Milano, M.T.; Marcus, K.J.; Yorke, E.; Hodgson, D.C.; Howell, R.M.; et al. Pediatric Normal Tissue Effects in the Clinic (PENTEC): An International Collaboration to Assess Normal Tissue Radiation Dose-Volume-Response Relationships for Children With Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2021, 31, 199–207. [Google Scholar] [CrossRef]
- Paulino, A.C.; Narayana, A.; Mohideen, M.N.; Jeswani, S. Posterior fossa boost in medulloblastoma: An analysis of dose to surrounding structures using 3-dimensional (conformal) radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 281–286. [Google Scholar] [CrossRef]
- Dunkel, I.J.; Gardner, S.L.; Garvin, J.H., Jr.; Goldman, S.; Shi, W.; Finlay, J.L. High-dose carboplatin, thiotepa, and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro-Oncology 2010, 12, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Milker-Zabel, S.; Zabel, A.; Thilmann, C.; Zuna, I.; Hoess, A.; Wannenmacher, M.; Debus, J. Results of three-dimensional stereotactically-guided radiotherapy in recurrent medulloblastoma. J. Neuro-Oncol. 2002, 60, 227–233. [Google Scholar] [CrossRef]
- Saran, F.; Baumert, B.G.; Creak, A.L.; Warrington, A.P.; Ashley, S.; Traish, D.; Brada, M. Hypofractionated stereotactic radiotherapy in the management of recurrent or residual medulloblastoma/PNET. Pediatr. Blood Cancer 2008, 50, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Gandola, L.; Spreafico, F.; Biassoni, V.; Luksch, R.; Collini, P.; Solero, C.N.; Simonetti, F.; Pignoli, E.; Cefalo, G.; et al. No salvage using high-dose chemotherapy plus/minus reirradiation for relapsing previously irradiated medulloblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1358–1363. [Google Scholar] [CrossRef]
- Bakst, R.L.; Dunkel, I.J.; Gilheeney, S.; Khakoo, Y.; Becher, O.; Souweidane, M.M.; Wolden, S.L. Reirradiation for recurrent medulloblastoma. Cancer 2011, 117, 4977–4982. [Google Scholar] [CrossRef]
- Tsang, D.S.; Sarhan, N.; Ramaswamy, V.; Nobre, L.; Yee, R.; Taylor, M.D.; Hawkins, C.; Bartels, U.; Huang, A.; Tabori, U.; et al. Re-irradiation for children with recurrent medulloblastoma in Toronto, Canada: A 20-year experience. J. Neuro-Oncol. 2019, 145, 107–114. [Google Scholar] [CrossRef]
- Gupta, T.; Maitre, M.; Sastri, G.J.; Krishnatry, R.; Shirsat, N.; Epari, S.; Sahay, A.; Chinnaswamy, G.; Patil, V.; Shetty, P.; et al. Outcomes of salvage re-irradiation in recurrent medulloblastoma correlate with age at initial diagnosis, primary risk-stratification, and molecular subgrouping. J. Neuro-Oncol. 2019, 144, 283–291. [Google Scholar] [CrossRef]
- Mayer, R.; Sminia, P. Reirradiation tolerance of the human brain. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 1350–1360. [Google Scholar] [CrossRef]
- Paul, S.; Sesikeran, B.N.; Patro, K.C.; Bhattacharya, K.; Palkonda, V.A.R. Re-irradiation in central nervous system tumors. J. Curr. Oncol. 2018, 1, 40–42. [Google Scholar] [CrossRef]
- Tsang, D.S.; Murray, L.; Ramaswamy, V.; Zapotocky, M.; Tabori, U.; Bartels, U.; Huang, A.; Dirks, P.B.; Taylor, M.D.; Hawkins, C.; et al. Craniospinal irradiation as part of re-irradiation for children with recurrent intracranial ependymoma. Neuro-Oncology 2019, 21, 547–557. [Google Scholar] [CrossRef]
- Tsang, D.S.; Laperriere, N.J. Re-irradiation for Paediatric Tumours. Clin. Oncol. 2019, 31, 191–198. [Google Scholar] [CrossRef]
- Patrice, S.J.; Tarbell, N.J.; Goumnerova, L.C.; Shrieve, D.C.; Black, P.M.; Loeffler, J.S. Results of radiosurgery in the management of recurrent and residual medulloblastoma. Pediatr. Neurosurg. 1995, 22, 197–203. [Google Scholar] [CrossRef]
- Raco, A.; Raimondi, A.J.; D’Alonzo, A.; Esposito, V.; Valentino, V. Radiosurgery in the management of pediatric brain tumors. Child’s Nerv. Syst. 2000, 16, 287–295. [Google Scholar] [CrossRef]
- Napieralska, A.; Braclik, I.; Radwan, M.; Mandera, M.; Blamek, S. Radiosurgery or hypofractionated stereotactic radiotherapy after craniospinal irradiation in children and adults with medulloblastoma and ependymoma. Child’s Nerv. Syst. 2019, 35, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, D.C.; Goumnerova, L.C.; Loeffler, J.S.; Dutton, S.; Black, P.M.; Alexander, E., III; Xu, R.; Kooy, H.; Silver, B.; Tarbell, N.J. Radiosurgery in the management of pediatric brain tumors. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 929–935. [Google Scholar] [CrossRef]
- Gultekin, M.; Cengiz, M.; Sezen, D.; Zorlu, F.; Yildiz, F.; Yazici, G.; Hurmuz, P.; Ozyigit, G.; Akyol, F.; Gurkaynak, M. Reirradiation of Pediatric Tumors Using Hypofractionated Stereotactic Radiotherapy. Technol. Cancer Res. Treat. 2017, 16, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.J.; Wuu, C.S. Radiation-induced second cancers: The impact of 3D-CRT and IMRT. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 83–88. [Google Scholar] [CrossRef]
- Dracham, C.B.; Shankar, A.; Madan, R. Radiation induced secondary malignancies: A review article. Radiat. Oncol. J. 2018, 36, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Powell, M.E. Intensity-modulated radiotherapy—What is it? Cancer Imaging 2004, 4, 68–73. [Google Scholar] [CrossRef] [Green Version]
- DeNunzio, N.J.; Yock, T.I. Modern Radiotherapy for Pediatric Brain Tumors. Cancers 2020, 12, 1533. [Google Scholar] [CrossRef]
- Bouffet, E.; Hawkins, C.E.; Ballourah, W.; Taylor, M.D.; Bartels, U.K.; Schoenhoff, N.; Tsangaris, E.; Huang, A.; Kulkarni, A.; Mabbot, D.J.; et al. Survival benefit for pediatric patients with recurrent ependymoma treated with reirradiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1541–1548. [Google Scholar] [CrossRef]
- Mascarin, M.; Dall’Oglio, S.; Palazzi, M.; Sartor, G.; Marradi, P.L.; Romano, M.; Maluta, S. A case of relapsed medulloblastoma treated with intensity-modulated radiotherapy and temozolomide. Tumori 2010, 96, 327–331. [Google Scholar] [CrossRef]
- de Rezende, A.C.P.; Weltman, E.; Chen, M.J.; Helito, J.K.; de Carvalho, I.T.; Sakuraba, R.K.; Silva, N.S.; Cappellano, A.M.; Hamerschlak, N. Intensity-modulated ventricular irradiation for intracranial germ-cell tumors: Survival analysis and impact of salvage re-irradiation. PLoS ONE 2019, 14, e0226350. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.R. Radiological use of fast protons. Radiology 1946, 47, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Grosshans, D. Proton therapy—Present and future. Adv. Drug Deliv. Rev. 2017, 109, 26–44. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.; Timmermann, B. Paediatric proton therapy. Br. J. Radiol. 2020, 93, 20190601. [Google Scholar] [CrossRef] [PubMed]
- DEGRO. Stellungnahme zur Strahlentherapie mit Protonen in Deutschland 2019 06/2020. Available online: https://www.degro.org/wp-content/uploads/2019/07/201907_StellungnahmeProtonen_final.pdf (accessed on 14 September 2020).
- Eaton, B.R.; Chowdhry, V.; Weaver, K.; Liu, L.; Ebb, D.; MacDonald, S.M.; Tarbell, N.J.; Yock, T.I. Use of proton therapy for re-irradiation in pediatric intracranial ependymoma. Radiother. Oncol. 2015, 116, 301–308. [Google Scholar] [CrossRef]
- Hill-Kayser, C.; Kirk, M. Brainstem-sparing craniospinal irradiation delivered with pencil beam scanning proton therapy. Pediatr. Blood Cancer 2015, 62, 718–720. [Google Scholar] [CrossRef]
- Lobon, M.J.; Bautista, F.; Riet, F.; Dhermain, F.; Canale, S.; Dufour, C.; Blauwblomme, T.; Zerah, M.; Beccaria, K.; Saint-Rose, C.; et al. Re-irradiation of recurrent pediatric ependymoma: Modalities and outcomes: A twenty-year survey. Springerplus 2016, 5, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latocha, K.; Frisch, S.; Tippelt, S.; Vermeren, X.; Errguibi, M.; Geismar, D.; Timmermann, B. Re-Bestrahlung mit Protonentherapie von Tumoren des zentralen Nervensystems: Ergebnisse aus den prospektiven Therapiestudien ProReg und KiProReg. Strahlenther. Onkol. 2021, 197, S73. [Google Scholar]
- Finlay, J.L.; Goldman, S.; Wong, M.C.; Cairo, M.; Garvin, J.; August, C.; Cohen, B.H.; Stanley, P.; Zimmerman, R.A.; Bostrom, B.; et al. Pilot study of high-dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children’s Cancer Group. J. Clin. Oncol. 1996, 14, 2495–2503. [Google Scholar] [CrossRef]
- Dunkel, I.J.; Boyett, J.M.; Yates, A.; Rosenblum, M.; Garvin, J.H., Jr.; Bostrom, B.C.; Goldman, S.; Sender, L.S.; Gardner, S.L.; Li, H.; et al. High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children’s Cancer Group. J. Clin. Oncol. 1998, 16, 222–228. [Google Scholar] [CrossRef]
- Mahoney, D.H., Jr.; Strother, D.; Camitta, B.; Bowen, T.; Ghim, T.; Pick, T.; Wall, D.; Yu, L.; Shuster, J.J.; Friedman, H. High-dose melphalan and cyclophosphamide with autologous bone marrow rescue for recurrent/progressive malignant brain tumors in children: A pilot pediatric oncology group study. J. Clin. Oncol. 1996, 14, 382–388. [Google Scholar] [CrossRef]
- Graham, M.L.; Herndon, J.E., II; Casey, J.R.; Chaffee, S.; Ciocci, G.H.; Krischer, J.P.; Kurtzberg, J.; Laughlin, M.J.; Longee, D.C.; Olson, J.F.; et al. High-dose chemotherapy with autologous stem-cell rescue in patients with recurrent and high-risk pediatric brain tumors. J. Clin. Oncol. 1997, 15, 1814–1823. [Google Scholar] [CrossRef]
- Busca, A.; Miniero, R.; Besenzon, L.; Cordero di Montezemolo, L.; Cenni, M.; Fagioli, F.; Sandri, A.; Vassallo, E.; Ricardi, U.; Madon, E. Etoposide-containing regimens with autologous bone marrow transplantation in children with malignant brain tumors. Child’s Nerv. Syst. 1997, 13, 572–577. [Google Scholar] [CrossRef]
- Gururangan, S.; Krauser, J.; Watral, M.A.; Driscoll, T.; Larrier, N.; Reardon, D.A.; Rich, J.N.; Quinn, J.A.; Vredenburgh, J.J.; Desjardins, A.; et al. Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro-Oncology 2008, 10, 745–751. [Google Scholar] [CrossRef]
- Shih, C.S.; Hale, G.A.; Gronewold, L.; Tong, X.; Laningham, F.H.; Gilger, E.A.; Srivastava, D.K.; Kun, L.E.; Gajjar, A.; Fouladi, M. High-dose chemotherapy with autologous stem cell rescue for children with recurrent malignant brain tumors. Cancer 2008, 112, 1345–1353. [Google Scholar] [CrossRef]
- Butturini, A.M.; Jacob, M.; Aguajo, J.; Vander-Walde, N.A.; Villablanca, J.; Jubran, R.; Erdreich-Epstein, A.; Marachelian, A.; Dhall, G.; Finlay, J.L. High-dose chemotherapy and autologous hematopoietic progenitor cell rescue in children with recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors: The impact of prior radiotherapy on outcome. Cancer 2009, 115, 2956–2963. [Google Scholar] [CrossRef]
- Cheuk, D.K.; Lee, T.L.; Chiang, A.K.; Ha, S.Y.; Chan, G.C. Autologous hematopoietic stem cell transplantation for high-risk brain tumors in children. J. Neuro-Oncol. 2008, 86, 337–347. [Google Scholar] [CrossRef]
- Fagioli, F.; Biasin, E.; Mastrodicasa, L.; Sandri, A.; Ferrero, I.; Berger, M.; Vassallo, E.; Madon, E. High-dose thiotepa and etoposide in children with poor-prognosis brain tumors. Cancer 2004, 100, 2215–2221. [Google Scholar] [CrossRef] [Green Version]
- Perez-Martinez, A.; Lassaletta, A.; Gonzalez-Vicent, M.; Sevilla, J.; Diaz, M.A.; Madero, L. High-dose chemotherapy with autologous stem cell rescue for children with high risk and recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors. J. Neuro-Oncol. 2005, 71, 33–38. [Google Scholar] [CrossRef]
- Valteau-Couanet, D.; Fillipini, B.; Benhamou, E.; Grill, J.; Kalifa, C.; Couanet, D.; Habrand, J.L.; Hartmann, O. High-dose busulfan and thiotepa followed by autologous stem cell transplantation (ASCT) in previously irradiated medulloblastoma patients: High toxicity and lack of efficacy. Bone Marrow Transplant. 2005, 36, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Osorio, D.S.; Dunkel, I.J.; Cervone, K.A.; Goyal, R.K.; Steve Lo, K.M.; Finlay, J.L.; Gardner, S.L. Tandem thiotepa with autologous hematopoietic cell rescue in patients with recurrent, refractory, or poor prognosis solid tumor malignancies. Pediatr. Blood Cancer 2018, 65, e26776. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.L.; Jacobsen, C.; Bunin, N.; Levine, J.; Goldman, F.; Bendel, A.; Joyce, M.; Anderson, P.; Rozans, M.; Wall, D.A.; et al. Phase I study of tandem high-dose chemotherapy with autologous peripheral blood stem cell rescue for children with recurrent brain tumors: A Pediatric Blood and MarrowTransplant Consortium study. Pediatr. Blood Cancer 2011, 57, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Gilheeney, S.W.; Khakoo, Y.; Souweidane, M.; Wolden, S.; Boulad, F.; Dunkel, I.J. Thiotepa/topotecan/carboplatin with autologous stem cell rescue in recurrent/refractory/poor prognosis pediatric malignancies of the central nervous system. Pediatr. Blood Cancer 2010, 54, 591–595. [Google Scholar] [CrossRef]
- Rosenfeld, A.; Kletzel, M.; Duerst, R.; Jacobsohn, D.; Haut, P.; Weinstein, J.; Rademaker, A.; Schaefer, C.; Evans, L.; Fouts, M.; et al. A phase II prospective study of sequential myeloablative chemotherapy with hematopoietic stem cell rescue for the treatment of selected high risk and recurrent central nervous system tumors. J. Neuro-Oncol. 2010, 97, 247–255. [Google Scholar] [CrossRef]
- Kadota, R.P.; Mahoney, D.H.; Doyle, J.; Duerst, R.; Friedman, H.; Holmes, E.; Kun, L.; Zhou, T.; Pollack, I.F. Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr. Blood Cancer 2008, 51, 675–678. [Google Scholar] [CrossRef] [Green Version]
- Egan, G.; Cervone, K.A.; Philips, P.C.; Belasco, J.B.; Finlay, J.L.; Gardner, S.L. Phase I study of temozolomide in combination with thiotepa and carboplatin with autologous hematopoietic cell rescue in patients with malignant brain tumors with minimal residual disease. Bone Marrow Transplant. 2016, 51, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Kang, J.; Yoo, K.H.; Sung, K.W.; Koo, H.H.; Lim, D.H.; Shin, H.J.; Kang, H.J.; Park, K.D.; Shin, H.Y.; et al. Efficacy of high-dose chemotherapy and autologous stem cell transplantation in patients with relapsed medulloblastoma: A report on the Korean Society for Pediatric Neuro-Oncology (KSPNO)-S-053 study. J. Korean Med. Sci. 2010, 25, 1160–1166. [Google Scholar] [CrossRef]
- Sung, K.W.; Yoo, K.H.; Cho, E.J.; Koo, H.H.; Lim, D.H.; Shin, H.J.; Ahn, S.D.; Ra, Y.S.; Choi, E.S.; Ghim, T.T. High-dose chemotherapy and autologous stem cell rescue in children with newly diagnosed high-risk or relapsed medulloblastoma or supratentorial primitive neuroectodermal tumor. Pediatr. Blood Cancer 2007, 48, 408–415. [Google Scholar] [CrossRef]
- Turner, C.D.; Gururangan, S.; Eastwood, J.; Bottom, K.; Watral, M.; Beason, R.; McLendon, R.E.; Friedman, A.H.; Tourt-Uhlig, S.; Miller, L.L.; et al. Phase II study of irinotecan (CPT-11) in children with high-risk malignant brain tumors: The Duke experience. Neuro-Oncology 2002, 4, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Hargrave, D.R.; Bouffet, E.; Gammon, J.; Tariq, N.; Grant, R.M.; Baruchel, S. Phase I study of fotemustine in pediatric patients with refractory brain tumors. Cancer 2002, 95, 1294–1301. [Google Scholar] [CrossRef]
- Kim, H.; Kang, H.J.; Lee, J.W.; Park, J.D.; Park, K.D.; Shin, H.Y.; Ahn, H.S. Irinotecan, vincristine, cisplatin, cyclophosphamide, and etoposide for refractory or relapsed medulloblastoma/PNET in pediatric patients. Child’s Nerv. Syst. 2013, 29, 1851–1858. [Google Scholar] [CrossRef]
- Aguilera, D.; Mazewski, C.; Fangusaro, J.; MacDonald, T.J.; McNall-Knapp, R.Y.; Hayes, L.L.; Kim, S.; Castellino, R.C. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: A multi-institutional experience. Child’s Nerv. Syst. 2013, 29, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Bonney, P.A.; Santucci, J.A.; Maurer, A.J.; Sughrue, M.E.; McNall-Knapp, R.Y.; Battiste, J.D. Dramatic response to temozolomide, irinotecan, and bevacizumab for recurrent medulloblastoma with widespread osseous metastases. J. Clin. Neurosci. 2016, 26, 161–163. [Google Scholar] [CrossRef]
- Aguilera, D.G.; Goldman, S.; Fangusaro, J. Bevacizumab and irinotecan in the treatment of children with recurrent/refractory medulloblastoma. Pediatr. Blood Cancer 2011, 56, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Schiavetti, A.; Varrasso, G.; Mollace, M.G.; Dominici, C.; Ferrara, E.; Papoff, P.; Di Biasi, C. Bevacizumab-containing regimen in relapsed/progressed brain tumors: A single-institution experience. Child’s Nerv. Syst. 2019, 35, 1007–1012. [Google Scholar] [CrossRef]
- Levy, A.S.; Krailo, M.; Chi, S.; Villaluna, D.; Springer, L.; Williams-Hughes, C.; Fouladi, M.; Gajjar, A. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr. Blood Cancer 2021, 68, e29031. [Google Scholar] [CrossRef]
- Zage, P.E. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Palmerini, E.; Jones, R.L.; Setola, E.; Picci, P.; Marchesi, E.; Luksch, R.; Grignani, G.; Cesari, M.; Longhi, A.; Abate, M.E.; et al. Irinotecan and temozolomide in recurrent Ewing sarcoma: An analysis in 51 adult and pediatric patients. Acta. Oncol. 2018, 57, 958–964. [Google Scholar] [CrossRef] [Green Version]
- Grill, J.; Geoerger, B.; Gesner, L.; Perek, D.; Leblond, P.; Canete, A.; Aerts, I.; Madero, L.; de Toledo Codina, J.S.; Verlooy, J.; et al. Phase II study of irinotecan in combination with temozolomide (TEMIRI) in children with recurrent or refractory medulloblastoma: A joint ITCC and SIOPE brain tumor study. Neuro-Oncology 2013, 15, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Di Giannatale, A.; Dias-Gastellier, N.; Devos, A.; Mc Hugh, K.; Boubaker, A.; Courbon, F.; Verschuur, A.; Ducassoul, S.; Malekzadeh, K.; Casanova, M.; et al. Phase II study of temozolomide in combination with topotecan (TOTEM) in relapsed or refractory neuroblastoma: A European Innovative Therapies for Children with Cancer-SIOP-European Neuroblastoma study. Eur. J. Cancer 2014, 50, 170–177. [Google Scholar] [CrossRef]
- Rubie, H.; Geoerger, B.; Frappaz, D.; Schmitt, A.; Leblond, P.; Ndiaye, A.; Aerts, I.; Le Deley, M.C.; Gentet, J.C.; Paci, A.; et al. Phase I study of topotecan in combination with temozolomide (TOTEM) in relapsed or refractory paediatric solid tumours. Eur. J. Cancer 2010, 46, 2763–2770. [Google Scholar] [CrossRef]
- Akyuz, C.; Demir, H.A.; Varan, A.; Yalcin, B.; Kutluk, T.; Buyukpamukcu, M. Temozolomide in relapsed pediatric brain tumors: 14 cases from a single center. Child’s Nerv. Syst. 2012, 28, 111–115. [Google Scholar] [CrossRef]
- Wang, C.H.; Hsu, T.R.; Wong, T.T.; Chang, K.P. Efficacy of temozolomide for recurrent embryonal brain tumors in children. Child’s Nerv. Syst. 2009, 25, 535–541. [Google Scholar] [CrossRef]
- Baruchel, S.; Diezi, M.; Hargrave, D.; Stempak, D.; Gammon, J.; Moghrabi, A.; Coppes, M.J.; Fernandez, C.V.; Bouffet, E. Safety and pharmacokinetics of temozolomide using a dose-escalation, metronomic schedule in recurrent paediatric brain tumours. Eur. J. Cancer 2006, 42, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, H.S.; Kretschmar, C.S.; Krailo, M.; Bernstein, M.; Kadota, R.; Fort, D.; Friedman, H.; Harris, M.B.; Tedeschi-Blok, N.; Mazewski, C.; et al. Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: A report from the Children’s Oncology Group. Cancer 2007, 110, 1542–1550. [Google Scholar] [CrossRef]
- Cefalo, G.; Massimino, M.; Ruggiero, A.; Barone, G.; Ridola, V.; Spreafico, F.; Potepan, P.; Abate, M.E.; Mascarin, M.; Garre, M.L.; et al. Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: An Italian multi-institutional phase II trial. Neuro-Oncology 2014, 16, 748–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, A.; Bakhshi, S. Metronomic chemotherapy in progressive pediatric malignancies: Old drugs in new package. Indian J. Pediatr. 2012, 79, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Butterfield, C.E.; Chaponis, D.M.; Laforme, A.M.; Folkman, J.; Kieran, M.W. Inhibition of tumor angiogenesis by oral etoposide. Exp. Ther. Med. 2010, 1, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Peyrl, A.; Chocholous, M.; Kieran, M.W.; Azizi, A.A.; Prucker, C.; Czech, T.; Dieckmann, K.; Schmook, M.T.; Haberler, C.; Leiss, U.; et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr. Blood Cancer 2012, 59, 511–517. [Google Scholar] [CrossRef]
- Ashley, D.M.; Meier, L.; Kerby, T.; Zalduondo, F.M.; Friedman, H.S.; Gajjar, A.; Kun, L.; Duffner, P.K.; Smith, S.; Longee, D. Response of recurrent medulloblastoma to low-dose oral etoposide. J. Clin. Oncol. 1996, 14, 1922–1927. [Google Scholar] [CrossRef]
- Davidson, A.; Gowing, R.; Lowis, S.; Newell, D.; Lewis, I.; Dicks-Mireaux, C.; Pinkerton, C.R. Phase II study of 21 day schedule oral etoposide in children. New Agents Group of the United Kingdom Children’s Cancer Study Group (UKCCSG). Eur. J. Cancer 1997, 33, 1816–1822. [Google Scholar] [CrossRef]
- Needle, M.N.; Molloy, P.T.; Geyer, J.R.; Herman-Liu, A.; Belasco, J.B.; Goldwein, J.W.; Sutton, L.; Phillips, P.C. Phase II study of daily oral etoposide in children with recurrent brain tumors and other solid tumors. Med. Pediatr. Oncol. 1997, 29, 28–32. [Google Scholar] [CrossRef]
- Perez-Somarriba, M.; Andion, M.; Lopez-Pino, M.A.; Lavarino, C.; Madero, L.; Lassaletta, A. Old drugs still work! Oral etoposide in a relapsed medulloblastoma. Child’s Nerv. Syst. 2019, 35, 865–869. [Google Scholar] [CrossRef]
- Ezoe, S. Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Environ. Res. Public Health 2012, 9, 2444–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Gou, P.; Dupret, J.M.; Chomienne, C.; Rodrigues-Lima, F. Etoposide, an anticancer drug involved in therapy-related secondary leukemia: Enzymes at play. Transl. Oncol. 2021, 14, 101169. [Google Scholar] [CrossRef]
- Slavc, I.; Peyrl, A.; Gojo, J.; Holm, S.; Blomgren, K.; Sehested, A.M.; Leblond, P.; Czech, T. MBCL-43. Recurrent Medulloblastoma—Long-term survival with a “MEMMAT” based antiangiogenic approach. Neuro-Oncology 2020, 22, iii397. [Google Scholar] [CrossRef]
- Sterba, J.; Valik, D.; Mudry, P.; Kepak, T.; Pavelka, Z.; Bajciova, V.; Zitterbart, K.; Kadlecova, V.; Mazanek, P. Combined biodifferentiating and antiangiogenic oral metronomic therapy is feasible and effective in relapsed solid tumors in children: Single-center pilot study. Onkologie 2006, 29, 308–313. [Google Scholar] [CrossRef]
- Zapletalova, D.; Andre, N.; Deak, L.; Kyr, M.; Bajciova, V.; Mudry, P.; Dubska, L.; Demlova, R.; Pavelka, Z.; Zitterbart, K.; et al. Metronomic chemotherapy with the COMBAT regimen in advanced pediatric malignancies: A multicenter experience. Oncology 2012, 82, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Chinnaswamy, G.; Sankaran, H.; Bhat, V.; Anand, K.C.; Saroha, M.; Prasad, M.; Vora, T.; Sahay, A.; Krishnatry, R.; Pungavkar, S.; et al. DEV-19. The role of COMBAT (Combined Oral Metronomic Bioifferentiating Antiangiogenic Areatment) in high-risk and relapsed medulloblastoma: A single institution experience. Neuro-Oncology 2018, 20 (Suppl. S2), i48–i49. [Google Scholar] [CrossRef]
- Ruggiero, A.; Rizzo, D.; Attina, G.; Lazzareschi, I.; Mastrangelo, S.; Maurizi, P.; Migliorati, R.; Bertolini, P.; Pastore, M.; Colosimo, C.; et al. Phase I study of temozolomide combined with oral etoposide in children with recurrent or progressive medulloblastoma. Eur. J. Cancer 2010, 46, 2943–2949. [Google Scholar] [CrossRef]
- Gajjar, A.; Pizer, B. Role of high-dose chemotherapy for recurrent medulloblastoma and other CNS primitive neuroectodermal tumors. Pediatr. Blood Cancer 2010, 54, 649–651. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Pereira, V.; Torrejon, J.; Kariyawasam, D.; Berlanga, P.; Guerrini-Rousseau, L.; Ayrault, O.; Varlet, P.; Tauziede-Espariat, A.; Puget, S.; Bolle, S.; et al. Clinical and molecular analysis of smoothened inhibitors in Sonic Hedgehog medulloblastoma. Neurooncol. Adv. 2021, 3, vdab097. [Google Scholar] [CrossRef]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkhuizen, T.; Reinders, M.G.; van Geel, M.; Hendriksen, A.J.; Paulussen, A.D.; Winnepenninckx, V.J.; Keymeulen, K.B.; Soetekouw, P.M.; van Steensel, M.A.; Mosterd, K. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2014, 71, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Petrirena, G.J.; Masliah-Planchon, J.; Sala, Q.; Pourroy, B.; Frappaz, D.; Tabouret, E.; Graillon, T.; Gentet, J.C.; Delattre, O.; Chinot, O.; et al. Recurrent extraneural sonic hedgehog medulloblastoma exhibiting sustained response to vismodegib and temozolomide monotherapies and inter-metastatic molecular heterogeneity at progression. Oncotarget 2018, 9, 10175–10183. [Google Scholar] [CrossRef] [Green Version]
- Ocasio, J.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019, 10, 5829. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, A.; Bowers, D.C.; Karajannis, M.A.; Leary, S.; Witt, H.; Gottardo, N.G. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J. Clin. Oncol. 2015, 33, 2986–2998. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sui, Y.; Li, Q.; Zhao, Y.; Dong, X.; Yang, J.; Liang, Z.; Han, Y.; Tang, Y.; Ma, J. Effective inhibition of MYC-amplified group 3 medulloblastoma by FACT-targeted curaxin drug CBL0137. Cell Death Dis. 2020, 11, 1029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, T.; Tian, S.; Meng, W.; Sui, Y.; Yang, J.; Wang, B.; Liang, Z.; Zhao, H.; Han, Y.; et al. Effective Inhibition of MYC-Amplified Group 3 Medulloblastoma Through Targeting EIF4A1. Cancer Manag. Res. 2020, 12, 12473–12485. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, N.K.; Kling, M.J.; Griggs, C.N.; Kesherwani, V.; Shukla, M.; McIntyre, E.M.; Ray, S.; Liu, Y.; McGuire, T.R.; Sharp, J.G.; et al. A Novel Combination Approach Targeting an Enhanced Protein Synthesis Pathway in MYC-driven (Group 3) Medulloblastoma. Mol. Cancer Ther. 2020, 19, 1351–1362. [Google Scholar] [CrossRef]
- Fouladi, M.; Park, J.R.; Stewart, C.F.; Gilbertson, R.J.; Schaiquevich, P.; Sun, J.; Reid, J.M.; Ames, M.M.; Speights, R.; Ingle, A.M.; et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: A Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 2010, 28, 3623–3629. [Google Scholar] [CrossRef] [Green Version]
- Muscal, J.A.; Thompson, P.A.; Horton, T.M.; Ingle, A.M.; Ahern, C.H.; McGovern, R.M.; Reid, J.M.; Ames, M.M.; Espinoza-Delgado, I.; Weigel, B.J.; et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: A Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr. Blood Cancer 2013, 60, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schuller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, S.; Gu, C.; Zhang, H.; Yu, C. Immune cell infiltration and cytokine secretion analysis reveal a non-inflammatory microenvironment of medulloblastoma. Oncol. Lett. 2020, 20, 397. [Google Scholar] [CrossRef]
- Murata, D.; Mineharu, Y.; Arakawa, Y.; Liu, B.; Tanji, M.; Yamaguchi, M.; Fujimoto, K.I.; Fukui, N.; Terada, Y.; Yokogawa, R.; et al. High programmed cell death 1 ligand-1 expression: Association with CD8+ T-cell infiltration and poor prognosis in human medulloblastoma. J. Neurosurg. 2018, 128, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, J.F.; Van Hecke, W.; Adriaansen, E.J.M.; Jansen, M.K.; Bouma, R.G.; Villacorta Hidalgo, J.; Fisch, P.; Broekhuizen, R.; Spliet, W.G.M.; Kool, M.; et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2018, 7, e1398877. [Google Scholar] [CrossRef]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.D.; Mitchell, D.A. Know your neighbors: Different tumor microenvironments have implications in immunotherapeutic targeting strategies across MB subgroups. Oncoimmunology 2016, 5, e1144002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.M.; Nirschl, C.J.; Polanczyk, M.J.; Bell, W.R.; Nirschl, T.R.; Harris-Bookman, S.; Phallen, J.; Hicks, J.; Martinez, D.; Ogurtsova, A.; et al. PD-L1 expression in medulloblastoma: An evaluation by subgroup. Oncotarget 2018, 9, 19177–19191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Varela-Guruceaga, M.; Tejada-Solis, S.; Garcia-Moure, M.; Fueyo, J.; Gomez-Manzano, C.; Patino-Garcia, A.; Alonso, M.M. Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors. Cancers 2018, 10, 226. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.M.; Brown, M.; Dobrikova, E.; Ramaswamy, V.; Taylor, M.D.; McLendon, R.; Sanks, J.; Chandramohan, V.; Bigner, D.; Gromeier, M. Poliovirus Receptor (CD155) Expression in Pediatric Brain Tumors Mediates Oncolysis of Medulloblastoma and Pleomorphic Xanthoastrocytoma. J. Neuropathol. Exp. Neurol. 2018, 77, 696–702. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Kreofsky, C.R.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Treatment of medulloblastoma with a modified measles virus. Neuro-Oncology 2010, 12, 1034–1042. [Google Scholar] [CrossRef]
- Lal, S.; Carrera, D.; Phillips, J.J.; Weiss, W.A.; Raffel, C. An oncolytic measles virus-sensitive Group 3 medulloblastoma model in immune-competent mice. Neuro-Oncology 2018, 20, 1606–1615. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Hutzen, B.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Oncolytic measles virus prolongs survival in a murine model of cerebral spinal fluid-disseminated medulloblastoma. Neuro-Oncology 2012, 14, 459–470. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.B.; Madsen, P.J.; Hegde, M.; Ahmed, N.; Cole, K.A.; Maris, J.M.; Resnick, A.C.; Storm, P.B.; Waanders, A.J. Immunotherapy for pediatric brain tumors: Past and present. Neuro-Oncology 2019, 21, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.F.; Kunos, C.A.; Villano, J.L.; Chauhan, A. Immunotherapy for Medulloblastoma: Current Perspectives. ImmunoTargets Ther. 2020, 9, 57–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.T.; Lin, W.Y.; Chen, Y.W.; Lin, C.F.; Wang, H.H.; Wu, S.H.; Lee, Y.Y. New Era of Immunotherapy in Pediatric Brain Tumors: Chimeric Antigen Receptor T-Cell Therapy. Int. J. Mol. Sci. 2021, 22, 2404. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.D.; Henson, J.C.; Breese, R.O.; Bielamowicz, K.J.; Rodriguez, A. CAR T Cell Therapy for Pediatric Brain Tumors. Front. Oncol. 2020, 10, 1582. [Google Scholar] [CrossRef]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Ode, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2020, 23, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Wei, J.; Liu, Y.; Wang, C.; Zhang, Y.; Tong, C.; Dai, G.; Wang, W.; Rasko, J.E.J.; Melenhorst, J.J.; Qian, W.; et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct. Target. Ther. 2020, 5, 134. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 2021, 397, 1010–1022. [Google Scholar] [CrossRef]
- North, R.J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 1982, 155, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Fleischhack, G.; Jaehde, U.; Bode, U. Pharmacokinetics following intraventricular administration of chemotherapy in patients with neoplastic meningitis. Clin. Pharmacokinet. 2005, 44, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Blasberg, R.G.; Patlak, C.; Fenstermacher, J.D. Intrathecal chemotherapy: Brain tissue profiles after ventriculocisternal perfusion. J. Pharmacol. Exp. Ther. 1975, 195, 73–83. [Google Scholar]
- Johanson, C.E.; Duncan, J.A.; Stopa, E.G.; Baird, A. Enhanced prospects for drug delivery and brain targeting by the choroid plexus-CSF route. Pharm. Res. 2005, 22, 1011–1037. [Google Scholar] [CrossRef]
- Shapiro, W.R.; Young, D.F.; Mehta, B.M. Methotrexate: Distribution in cerebrospinal fluid after intravenous, ventricular and lumbar injections. N. Engl. J. Med. 1975, 293, 161–166. [Google Scholar] [CrossRef]
- Blaney, S.M.; Poplack, D.G.; Godwin, K.; McCully, C.L.; Murphy, R.; Balis, F.M. Effect of body position on ventricular CSF methotrexate concentration following intralumbar administration. J. Clin. Oncol. 1995, 13, 177–179. [Google Scholar] [CrossRef]
- Bode, U.; Magrath, I.T.; Bleyer, W.A.; Poplack, D.G.; Glaubiger, D.L. Active transport of methotrexate from cerebrospinal fluid in humans. Cancer Res. 1980, 40, 2184–2187. [Google Scholar] [PubMed]
- Mynarek, M.; von Hoff, K.; Pietsch, T.; Ottensmeier, H.; Warmuth-Metz, M.; Bison, B.; Pfister, S.; Korshunov, A.; Sharma, T.; Jaeger, N.; et al. Nonmetastatic Medulloblastoma of Early Childhood: Results From the Prospective Clinical Trial HIT-2000 and An Extended Validation Cohort. J. Clin. Oncol. 2020, 38, 2028–2040. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Tippelt, S.; Siegler, N.; Reichling, S.; Zimmermann, M.; Mikasch, R.; Bode, U.; Gnekow, A.; Pietsch, T.; Benesch, M.; et al. Intraventricular etoposide safety and toxicity profile in children and young adults with refractory or recurrent malignant brain tumors. J. Neuro-Oncol. 2016, 128, 463–471. [Google Scholar] [CrossRef]
- Pompe, R.S.; von Bueren, A.O.; Mynarek, M.; von Hoff, K.; Friedrich, C.; Kwiecien, R.; Treulieb, W.; Lindow, C.; Deinlein, F.; Fleischhack, G.; et al. Intraventricular methotrexate as part of primary therapy for children with infant and/or metastatic medulloblastoma: Feasibility, acute toxicity and evidence for efficacy. Eur. J. Cancer 2015, 51, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Mack, F.; Baumert, B.G.; Schafer, N.; Hattingen, E.; Scheffler, B.; Herrlinger, U.; Glas, M. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat. Rev. 2016, 43, 83–91. [Google Scholar] [CrossRef]
- Bleyer, W.A.; Poplack, D.G.; Simon, R.M. “Concentration x time” methotrexate via a subcutaneous reservoir: A less toxic regimen for intraventricular chemotherapy of central nervous system neoplasms. Blood 1978, 51, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimm, S.; Collins, J.M.; Miser, J.; Chatterji, D.; Poplack, D.G. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984, 35, 826–830. [Google Scholar] [CrossRef]
- Blaney, S.M.; Tagen, M.; Onar-Thomas, A.; Berg, S.L.; Gururangan, S.; Scorsone, K.; Su, J.; Goldman, S.; Kieran, M.W.; Kun, L.; et al. A phase-1 pharmacokinetic optimal dosing study of intraventricular topotecan for children with neoplastic meningitis: A Pediatric Brain Tumor Consortium study. Pediatr. Blood Cancer 2013, 60, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischhack, G.; Reif, S.; Hasan, C.; Jaehde, U.; Hettmer, S.; Bode, U. Feasibility of intraventricular administration of etoposide in patients with metastatic brain tumours. Br. J. Cancer 2001, 84, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, N.; Mori, T.; Kawashima, H.; Fujiki, M.; Abe, T.; Kaku, T.; Konisi, Y.; Takeyama, M.; Hori, S. Pharmacokinetics of nimustine, methotrexate, and cytosine arabinoside during cerebrospinal fluid perfusion chemotherapy in patients with disseminated brain tumors. Eur. J. Clin. Pharmacol. 1998, 54, 415–420. [Google Scholar] [CrossRef]
- Roth, P.; Weller, M. Management of neoplastic meningitis. Chin. Clin. Oncol. 2015, 4, 26. [Google Scholar]
- Bleyer, A.; Choi, M.; Wang, S.J.; Fuller, C.D.; Raney, R.B. Increased vulnerability of the spinal cord to radiation or intrathecal chemotherapy during adolescence: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2009, 53, 1205–1210. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Yeung, D.Y.; Chan, J.C. Intrathecal chemotherapy for hematologic malignancies: Drugs and toxicities. Ann. Hematol. 2009, 88, 193–201. [Google Scholar] [CrossRef]
- Montejo, C.; Navarro-Otano, J.; Maya-Casalprim, G.; Campolo, M.; Casanova-Molla, J. Acute lumbar polyradiculoneuropathy as early sign of methotrexate intrathecal neurotoxicity: Case report and literature review. Clin. Case Rep. 2019, 7, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Yoon, J.Y.; Park, H.J.; Son, M.H.; Kim, S.H.; Kim, W.; Kim, H.J.; Lee, S.H.; Park, B.K. Diffuse cerebral vasospasm with infarct after intrathecal cytarabine in childhood leukemia. Pediatr. Int. 2014, 56, 921–924. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Humm, J.L.; Zanzonico, P.B.; Haque, S.; Dunkel, I.J.; Wolden, S.L.; Donzelli, M.; Goldman, D.A.; Lewis, J.S.; et al. A phase II study of radioimmunotherapy with intraventricular (131) I-3F8 for medulloblastoma. Pediatr. Blood Cancer 2018, 65, e26754. [Google Scholar] [CrossRef]
- Baenziger, P.H.; Moody, K. Palliative Care for Children with Central Nervous System Malignancies. Bioengineering 2018, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Bounajem, M.T.; Karsy, M.; Jensen, R.L. Liquid biopsies for the diagnosis and surveillance of primary pediatric central nervous system tumors: A review for practicing neurosurgeons. Neurosurg. Focus 2020, 48, E8. [Google Scholar] [CrossRef] [Green Version]
- Madlener, S.; Gojo, J. Liquid Biomarkers for Pediatric Brain Tumors: Biological Features, Advantages and Perspectives. J. Pers. Med. 2020, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Gardner, S.; Snuderl, M. The Role of Liquid Biopsies in Pediatric Brain Tumors. J. Neuropathol. Exp. Neurol. 2020, 79, 934–940. [Google Scholar] [CrossRef]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta. Neuropathol. Commun. 2017, 5, 28. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Smith, K.S.; Kumar, R.; Paul, L.; Bihannic, L.; Lin, T.; Maass, K.K.; Pajtler, K.W.; Chintagumpala, M.; Su, J.M.; et al. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021, 39, 1519–1530.e4. [Google Scholar] [CrossRef]
- Li, J.; Zhao, S.; Lee, M.; Yin, Y.; Li, J.; Zhou, Y.; Ballester, L.Y.; Esquenazi, Y.; Dashwood, R.H.; Davies, P.J.A.; et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 2020, 6, eabb5427. [Google Scholar] [CrossRef]
- Escudero, L.; Llort, A.; Arias, A.; Diaz-Navarro, A.; Martinez-Ricarte, F.; Rubio-Perez, C.; Mayor, R.; Caratu, G.; Martinez-Saez, E.; Vazquez-Mendez, E.; et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 2020, 11, 5376. [Google Scholar] [CrossRef]
- Braoudaki, M.; Lambrou, G.I.; Giannikou, K.; Milionis, V.; Stefanaki, K.; Birks, D.K.; Prodromou, N.; Kolialexi, A.; Kattamis, A.; Spiliopoulou, C.A.; et al. Microrna expression signatures predict patient progression and disease outcome in pediatric embryonal central nervous system neoplasms. J. Hematol. Oncol. 2014, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Azzarelli, R. Organoid Models of Glioblastoma to Study Brain Tumor Stem Cells. Front. Cell Dev. Biol. 2020, 8, 220. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Sanden, E.; Dyberg, C.; Krona, C.; Gallo-Oller, G.; Olsen, T.K.; Enriquez Perez, J.; Wickstrom, M.; Estekizadeh, A.; Kool, M.; Visse, E.; et al. Establishment and characterization of an orthotopic patient-derived Group 3 medulloblastoma model for preclinical drug evaluation. Sci. Rep. 2017, 7, 46366. [Google Scholar] [CrossRef] [Green Version]
- Cook Sangar, M.L.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Brabetz, S.; Leary, S.E.S.; Grobner, S.N.; Nakamoto, M.W.; Seker-Cin, H.; Girard, E.J.; Cole, B.; Strand, A.D.; Bloom, K.L.; Hovestadt, V.; et al. A biobank of patient-derived pediatric brain tumor models. Nat. Med. 2018, 24, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Rusert, J.M.; Juarez, E.F.; Brabetz, S.; Jensen, J.; Garancher, A.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Wahab, S.; Udaka, Y.T.; Finlay, D.; et al. Functional Precision Medicine Identifies New Therapeutic Candidates for Medulloblastoma. Cancer Res. 2020, 80, 5393–5407. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Essential Sequences: 1.5 Tesla Scanner | |||
|---|---|---|---|
| Sequence | Technique | Parameters | Plane |
| T1W | 2D SE, TSE/FSE | Slice thickness ≤4 mm | Axial (along AC-PC axis) |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T2W | 2D SE, TSE/FSE | Slice thickness ≤4 mm | Axial |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T2 FLAIR | 2D TSE/FSE | Slice thickness ≤4 mm | Axial or coronal |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T1W + contrast | 2D SE, TSE/FSE | Slice thickness ≤4 mm | Axial, coronal and sagittal |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| DWI with ADC | 2D EPI | Slice thickness ≤4 mm | Axial |
| Slice gap ≤1 mm (10% of slice thickness) | |||
| b = 0 and 1000. ADC maps reconstructed on-line | |||
| Essential Sequences: 3 Tesla Scanner | |||
| Sequence | Technique | Parameters | Plane |
| T1W | 3D gradient echo (MP-RAGE/IR-SPGR/Fast SPGR/3D TFE/3D FFE) | Slice thickness ≤1 mm with no slice gap | Axial or sagittal |
| Isotropic voxel resolution of 1 mm × 1 mm × 1 mm desirable | |||
| T2W | 2D SE, TSE/FSE | Slice thickness ≤4 mm | Axial |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T2 FLAIR | 2D TSE/FSE | Slice thickness ≤4 mm | Axial or coronal |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T1W + contrast | 2D SE, TSE/FSE | Slice thickness ≤4 mm | Axial |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| T1W + contrast | 3D gradient echo (MP-RAGE/IR-SPGR/Fast SPGR/3D TFE/3D FFE) | Slice thickness ≤1 mm with no slice gap | Axial or sagittal, to match pre-contrast |
| Isotropic voxel resolution of 1 mm × 1 mm × 1 mm desirable | |||
| DWI with ADC | 2D EPI | Slice thickness ≤4 mm | Axial |
| Slice gap ≤1 mm (10% of slice thickness desirable) | |||
| b = 0 and 1000, ADC maps reconstructed on-line | |||
| Resolution parameters: Field of view—230 mm (range 220–250 mm depending on head size). Matrix size—minimum 256 (512 is desirable for better resolution; 96–128 for EPI sequences). | |||
| Optional Sequences | |||
| Sequence | Technique | Parameters | Plane |
| T1W | 3D gradient echo (on 1.5 T)/3D T1 TSE | - | Axial or sagittal |
| T2 FLAIR | 3D gradient echo * | - | Axial or sagittal |
| Heavily weighted T2W | 2D or 3D CISS/bFFE/FIESTA ** | - | Axial or coronal or sagittal |
| Advanced MRI | DTI, perfusion and spectroscopy | - | NA |
| Essential Sequences | |||
|---|---|---|---|
| Sequence | Technique | Parameters | Plane |
| T1W + contrast | 2D SE/TSE | Slice thickness ≤3 mmSlice gap <0.5 mm | Sagittal whole spine (entire dural sac) |
| T1W + contrast | 2D SE/TSE or 3D gradient | Slice thickness 4–5 mmNo slice gap | Axial—suspicious areas * |
| Matrix size—Minimum 256 (512 is desirable for better resolution). | |||
| Optional Sequences | |||
| Sequence | Technique | Parameters | Plane |
| T2W | 2D SE/TSE | - | Sagittal whole spine |
| T2W | 2D SE/TSE | - | Axial—suspicious areas |
| Heavily weighted T2W | 2D or 3D CISS/bFFE/FIESTA ** | - | Sagittal ± axial |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, R.M.; Plasschaert, S.L.A.; Timmermann, B.; Dufour, C.; Aquilina, K.; Avula, S.; Donovan, L.; Lequin, M.; Pietsch, T.; Thomale, U.; et al. Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers 2022, 14, 126. https://doi.org/10.3390/cancers14010126
Hill RM, Plasschaert SLA, Timmermann B, Dufour C, Aquilina K, Avula S, Donovan L, Lequin M, Pietsch T, Thomale U, et al. Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers. 2022; 14(1):126. https://doi.org/10.3390/cancers14010126
Chicago/Turabian StyleHill, Rebecca M., Sabine L. A. Plasschaert, Beate Timmermann, Christelle Dufour, Kristian Aquilina, Shivaram Avula, Laura Donovan, Maarten Lequin, Torsten Pietsch, Ulrich Thomale, and et al. 2022. "Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment" Cancers 14, no. 1: 126. https://doi.org/10.3390/cancers14010126
APA StyleHill, R. M., Plasschaert, S. L. A., Timmermann, B., Dufour, C., Aquilina, K., Avula, S., Donovan, L., Lequin, M., Pietsch, T., Thomale, U., Tippelt, S., Wesseling, P., Rutkowski, S., Clifford, S. C., Pfister, S. M., Bailey, S., & Fleischhack, G. (2022). Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers, 14(1), 126. https://doi.org/10.3390/cancers14010126