
Aerobic Exercise Training and In Vivo Akt Activation Counteract Cancer Cachexia by Inducing a Hypertrophic Profile through eIF-2α Modulation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Physical Exercise
2.2.1. Maximal Running Capacity Test (MRC)
2.2.2. Aerobic Exercise Training (AET)
2.2.3. Single Exercise Session
2.3. Adult Skeletal Muscle In Vivo Electrotransfer and Plasmids Constructs
2.4. Muscle Morphology and Quantitative Analyses
2.5. Immunostaining
2.6. Western Blot Analyses
2.7. Antibodies for Western Blot and Immunostaining
2.8. Muscle Mechanics
2.8.1. In Vivo
2.8.2. In Situ
2.9. Measurements of In Vivo Protein Synthesis
2.10. Proteasome Activity
2.11. Statistics
3. Results
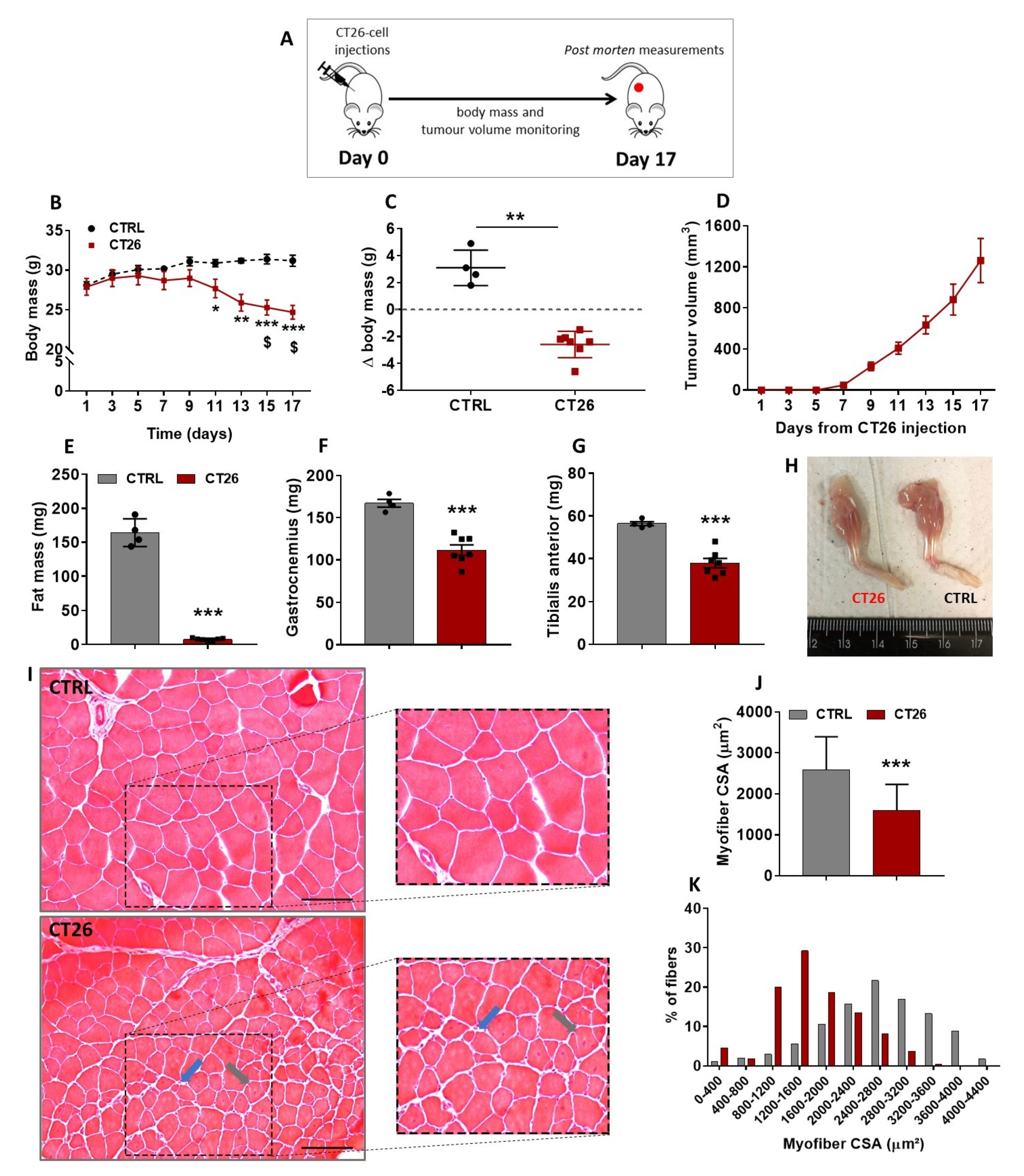
3.1. Murine Colon Adenocarcinoma Cell Line, CT26, Induces Severe Cachexia in Mice
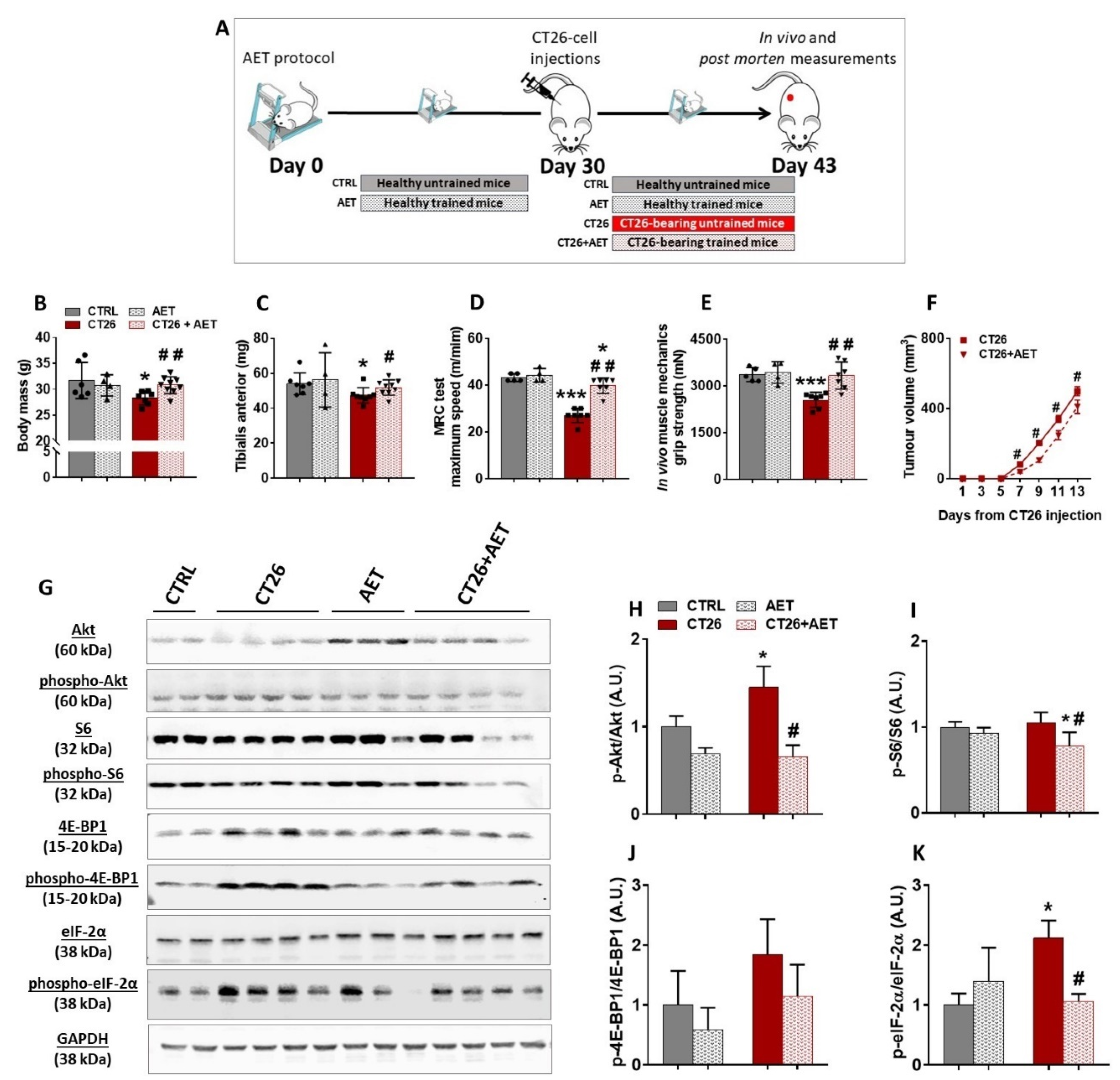
3.2. Aerobic Exercise Training Delays Tumor Progression, Prevents Cachexia, Modulates Akt/mTORC1 Signaling, and Suggests Eukaryotic Initiation Factor-2α as a Target Involved in the Skeletal Muscle Plasticity in Cancer Context
3.3. Akt-Induced Hypertrophy Is Necessary to Prevent and to Revert Cancer-Associated Muscle Wasting
3.4. Akt-Related Hypertrophy Rescues Muscle Force and Induces a Better Translation Initiation Process in Cachectic Muscles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ni, J.; Zhang, L. Cancer Cachexia: Definition, Staging, and Emerging Treatments. Cancer Manag. Res. 2020, 12, 5597–5605. [Google Scholar] [CrossRef]
- Lena, A.; Anker, M.S.; Springer, J. Muscle Wasting and Sarcopenia in Heart Failure—The Current State of Science. Int. J. Mol. Sci. 2020, 21, 6549. [Google Scholar] [CrossRef]
- Wang, M.; Wu, X.; Gan, L.; Teng, Z.; Zhang, H.; Zhang, Y. Overexpression of Dnmt3a ameliorates diabetic muscle atrophy by modulating the Pten/Akt pathway. Exp. Physiol. 2020, 105, 1918–1927. [Google Scholar] [CrossRef]
- Cao, R.Y.; Li, J.; Dai, Q.; Li, Q.; Yang, J. Muscle Atrophy: Present and Future. In Muscle Atrophy; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; Volume 1088, pp. 605–624. [Google Scholar] [CrossRef]
- Farkas, J.; von Haehling, S.; Kalantar-Zadeh, K.; Morley, J.E.; Anker, S.D.; Lainscak, M. Cachexia as a major public health problem: Frequent, costly, and deadly. J. Cachexia Sarcopenia Muscle 2013, 4, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedenreich, C.M.; Cust, E.A. Physical activity and breast cancer risk: Impact of timing, type and dose of activity and population subgroup effects. Br. J. Sports Med. 2008, 42, 636–647. [Google Scholar] [CrossRef]
- Neilson, H.K.; Friedenreich, C.M.; Brockton, N.T.; Millikan, R.C. Physical activity and postmeno-pausal breast cancer: Proposed biologic mechanisms and areas for future research. Cancer Epidemiol. Biomark. Prev. 2009, 18, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Diaz, M.B. Cancer Cachexia: More Than Skeletal Muscle Wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic exercise and pharmacological treatments counteract ca-chexia by modulating autophagy in colon cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.R.R.; Neves, W.D.; de Almeida, N.R.; Eichelberger, E.J.; Jannig, P.R.; Voltarelli, V.A.; Tobi-as, G.C.; Bechara, L.R.G.; de Paula Faria, D.; Alves, M.J.N.; et al. Exercise training reverses cancer-induced oxidative stress and decrease in muscle cops2/trip15/alien. Mol. Metab. 2020, 39, 101012. [Google Scholar] [CrossRef]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms Modulating Skeletal Muscle Phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.G.; Dyar, K.A.; Nogara, L.; Solagna, F.; Marabita, M.; Baraldo, M.; Chemello, F.; Germi-nario, E.; Romanello, V.; Nolte, H.; et al. Comparative analysis of muscle hypertrophy models re-veals divergent gene transcription profiles and points to translational regulation of muscle growth through increased mtor signaling. Front Physiol. 2017, 8, 968. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2015, 54, 11–19. [Google Scholar] [CrossRef]
- Pallafacchina, G.; Calabria, E.; Serrano, A.L.; Kalhovde, J.M.; Schiaffino, S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl. Acad. Sci. USA 2002, 99, 9213–9218. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated ca-chexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both peri-cytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Investig. 2003, 111, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.; Rolim, N.P.; Bartholomeu, J.B.; Gobatto, C.A.; Kokubun, E.; Brum, P.C. Maximal lac-tate steady state in running mice: Effect of exercise training. Clin. Exp. Pharmacol. Physiol. 2007, 34, 760–765. [Google Scholar] [CrossRef]
- Bacurau, A.V.N.; Belmonte, M.A.; Navarro, F.; Moraes, M.R.; Pontes, J.F.L.; Pesquero, J.; Araujo, R.; Bacurau, R.F.P. Effect of a High-Intensity Exercise Training on the Metabolism and Function of Macrophages and Lymphocytes of Walker 256 Tumor–Bearing Rats. Exp. Biol. Med. 2007, 232, 1289–1299. [Google Scholar] [CrossRef]
- Bacurau, A.V.; Jannig, P.R.; de Moraes, W.M.; Cunha, T.F.; Medeiros, A.; Barberi, L.; Coelho, M.A.; Bacurau, R.F.; Ugrinowitsch, C.; Musarò, A.; et al. Akt/mTOR pathway contributes to skeletal muscle anti-atrophic effect of aerobic exercise training in heart failure mice. Int. J. Cardiol. 2016, 214, 137–147. [Google Scholar] [CrossRef]
- Bueno, C.R., Jr.; Ferreira, J.C.; Pereira, M.G.; Bacurau, A.V.; Brum, P.C. Aerobic exercise training improves skeletal muscle function and Ca2+ handling-related protein expression in sympathetic hy-peractivity-induced heart failure. J. Appl. Physiol. (1985) 2010, 109, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, T.F.; Moreira, J.B.N.; Paixão, N.A.; Campos, J.C.; Monteiro, A.W.A.; Bacurau, A.V.N.; Bueno, C.R.; Ferreira, J.C.B.; Brum, P.C. Aerobic exercise training upregulates skeletal muscle calpain and ubiquitin-proteasome systems in healthy mice. J. Appl. Physiol. 2012, 112, 1839–1846. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.G.; Ferreira, J.C.B.; BuenoJr, C.R.; Mattos, K.C.; Rosa, K.T.; Irigoyen, M.C.; Oliveira, E.M.; Krieger, J.E.; Brum, P.C. Exercise training reduces cardiac angiotensin II levels and prevents cardiac dysfunction in a genetic model of sympathetic hyperactivity-induced heart failure in mice. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 105, 843–850. [Google Scholar] [CrossRef]
- Rolim, N.P.; Medeiros, A.; Rosa, K.T.; Mattos, K.C.; Irigoyen, M.C.; Krieger, E.M.; Krieger, J.E.; Negrao, C.E.; Brum, P.C. Exercise training improves the net balance of cardiac Ca2+ handling protein expression in heart failure. Physiol. Genomics 2007, 29, 246–252. [Google Scholar] [CrossRef]
- Vieira, J.; Cunha, T.; Paixão, N.; Dourado, P.; Carrascoza, L.; Bacurau, A.; Brum, P. Exercise intolerance establishment in pulmonary hypertension: Preventive effect of aerobic exercise training. Life Sci. 2020, 261, 118298. [Google Scholar] [CrossRef]
- Donà, M.; Sandri, M.; Rossini, K.; Dell’Aica, I.; Podhorska-Okolow, M.; Carraro, U. Functional in vivo gene transfer into the myofibers of adult skeletal muscle. Biochem. Biophys. Res. Commun. 2003, 312, 1132–1138. [Google Scholar] [CrossRef]
- Smith, L.R.; Barton, E.R. SMASH—Semi-automatic muscle analysis using segmentation of histology: A MATLAB application. Skelet. Muscle 2014, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.G.; Baptista, I.L.; Carlassara, E.O.C.; Moriscot, A.S.; Aoki, M.S.; Miyabara, E.H. Leucine Supplementation Improves Skeletal Muscle Regeneration after Cryolesion in Rats. PLoS ONE 2014, 9, e85283. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.G.; Silva, M.T.; Carlassara, E.O.C.; Gonçalves, D.A.; Abrahamsohn, P.A.; Kettelhut, I.C.; Moriscot, A.S.; Aoki, M.S.; Miyabara, E.H. Leucine Supplementation Accelerates Connective Tissue Repair of Injured Tibialis Anterior Muscle. Nutrients 2014, 6, 3981–4001. [Google Scholar] [CrossRef] [Green Version]
- Blaauw, B.; Canato, M.; Agatea, L.; Toniolo, L.; Mammucari, C.; Masiero, E.; Abraham, R.; Sandri, M.; Schiaffino, S.; Reggiani, C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009, 23, 3896–3905. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Hornberger, T.A. Measuring protein synthesis with sunset: A valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 2013, 41, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetto, A.; Rupert, J.E.; Barreto, R.; Zimmers, T.A. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp. 2016, 30, 54893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K.; Saltin, B. Exercise as medicine–Evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. 3), 1–72. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.W.; Eves, N.D.; Scott, J.M. Bench-to-bedside approaches for personalized exercise therapy in cancer. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 684–694. [Google Scholar] [CrossRef]
- Wen, Y.; Alimov, A.P.; McCarthy, J.J. Ribosome biogenesis is necessary for skeletal muscle hyper-trophy. Exerc. Sport Sci. Rev. 2016, 44, 110–115. [Google Scholar] [CrossRef]
- Marabita, M.; Baraldo, M.; Solagna, F.; Ceelen, J.J.; Sartori, R.; Nolte, H.; Nemazanyy, I.; Pyronnet, S.; Kruger, M.; Pende, M.; et al. S6k1 is required for increasing skeletal muscle force during hyper-trophy. Cell Rep. 2016, 17, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mtor-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Klann, K.; Tascher, G.; Münch, C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α. Mol. Cell 2019, 77, 913–925.e4. [Google Scholar] [CrossRef] [Green Version]
- Khamoui, A.; Park, B.-S.; Kim, D.-H.; Yeh, M.-C.; Oh, S.-L.; Elam, M.L.; Jo, E.; Arjmandi, B.H.; Salazar, G.; Grant, S.; et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism 2016, 65, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Baraldo, M.; Geremia, A.; Pirazzini, M.; Nogara, L.; Solagna, F.; Türk, C.; Nolte, H.; Romanello, V.; Megighian, A.; Boncompagni, S.; et al. Skeletal muscle mTORC1 regulates neuromuscular junction stability. J. Cachexia Sarcopenia Muscle 2019, 11, 208–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.C.; Lee, I.-M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; De Gonzalez, A.B.; Hartge, P.; et al. Association of Leisure-Time Physical Activity with Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef]
- Ding, D.; Kolbe-Alexander, T.; Nguyen, B.; Katzmarzyk, P.T.; Pratt, M.; Lawson, K.D. The econom-ic burden of physical inactivity: A systematic review and critical appraisal. Br. J. Sports Med. 2017, 51, 1392–1409. [Google Scholar] [CrossRef] [Green Version]
- Orr-Asman, M.A.; Chu, Z.; Jiang, M.; Worley, M.; LaSance, K.; Koch, S.E.; Carreira, V.S.; Dahche, H.M.; Plas, D.R.; Komurov, K.; et al. mTOR Kinase Inhibition Effectively Decreases Progression of a Subset of Neuroendocrine Tumors that Progress on Rapalog Therapy and Delays Cardiac Impairment. Mol. Cancer Ther. 2017, 16, 2432–2441. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, E.; Manning, B.D. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Valvezan, A.J.; Manning, B.D. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab. 2019, 1, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Eley, H.L.; Skipworth, R.J.; Deans, D.A.; Fearon, K.C.; Tisdale, M.J. Increased expression of phos-phorylated forms of rna-dependent protein kinase and eukaryotic initiation factor 2alpha may sig-nal skeletal muscle atrophy in weight-losing cancer patients. Br. J. Cancer 2008, 98, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Ishihara, H.; Yamada, T.; Tamura, A.; Usui, M.; Tominaga, R.; Munakata, Y.; Satake, C.; Katagiri, H.; Tashiro, F.; et al. Atf4-mediated induction of 4e-bp1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab. 2008, 7, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Shan, B.; Lee, B.-H.; Zhu, K.; Zhang, T.; Sun, H.; Liu, M.; Shi, L.; Liang, W.; Qian, L.; et al. Author response: Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. ELife 2015, 4, e10510. [Google Scholar] [CrossRef] [PubMed]
- Zismanov, V.; Chichkov, V.; Colangelo, V.; Jamet, S.; Wang, S.; Syme, A.; Koromilas, A.E.; Crist, C. Phosphorylation of eif2alpha is a translational control mechanism regulating muscle stem cell qui-escence and self-renewal. Cell Stem Cell 2016, 18, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of murf1/murf2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.G.; Voltarelli, V.A.; Tobias, G.C.; de Souza, L.; Borges, G.S.; Paixão, A.O.; de Almeida, N.R.; Bowen, T.S.; Demasi, M.; Miyabara, E.H.; et al. Aerobic Exercise Training and In Vivo Akt Activation Counteract Cancer Cachexia by Inducing a Hypertrophic Profile through eIF-2α Modulation. Cancers 2022, 14, 28. https://doi.org/10.3390/cancers14010028
Pereira MG, Voltarelli VA, Tobias GC, de Souza L, Borges GS, Paixão AO, de Almeida NR, Bowen TS, Demasi M, Miyabara EH, et al. Aerobic Exercise Training and In Vivo Akt Activation Counteract Cancer Cachexia by Inducing a Hypertrophic Profile through eIF-2α Modulation. Cancers. 2022; 14(1):28. https://doi.org/10.3390/cancers14010028
Chicago/Turabian StylePereira, Marcelo G., Vanessa A. Voltarelli, Gabriel C. Tobias, Lara de Souza, Gabriela S. Borges, Ailma O. Paixão, Ney R. de Almeida, Thomas Scott Bowen, Marilene Demasi, Elen H. Miyabara, and et al. 2022. "Aerobic Exercise Training and In Vivo Akt Activation Counteract Cancer Cachexia by Inducing a Hypertrophic Profile through eIF-2α Modulation" Cancers 14, no. 1: 28. https://doi.org/10.3390/cancers14010028
APA StylePereira, M. G., Voltarelli, V. A., Tobias, G. C., de Souza, L., Borges, G. S., Paixão, A. O., de Almeida, N. R., Bowen, T. S., Demasi, M., Miyabara, E. H., & Brum, P. C. (2022). Aerobic Exercise Training and In Vivo Akt Activation Counteract Cancer Cachexia by Inducing a Hypertrophic Profile through eIF-2α Modulation. Cancers, 14(1), 28. https://doi.org/10.3390/cancers14010028