
Near-Haploidy and Low-Hypodiploidy in B-Cell Acute Lymphoblastic Leukemia: When Less Is Too Much

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
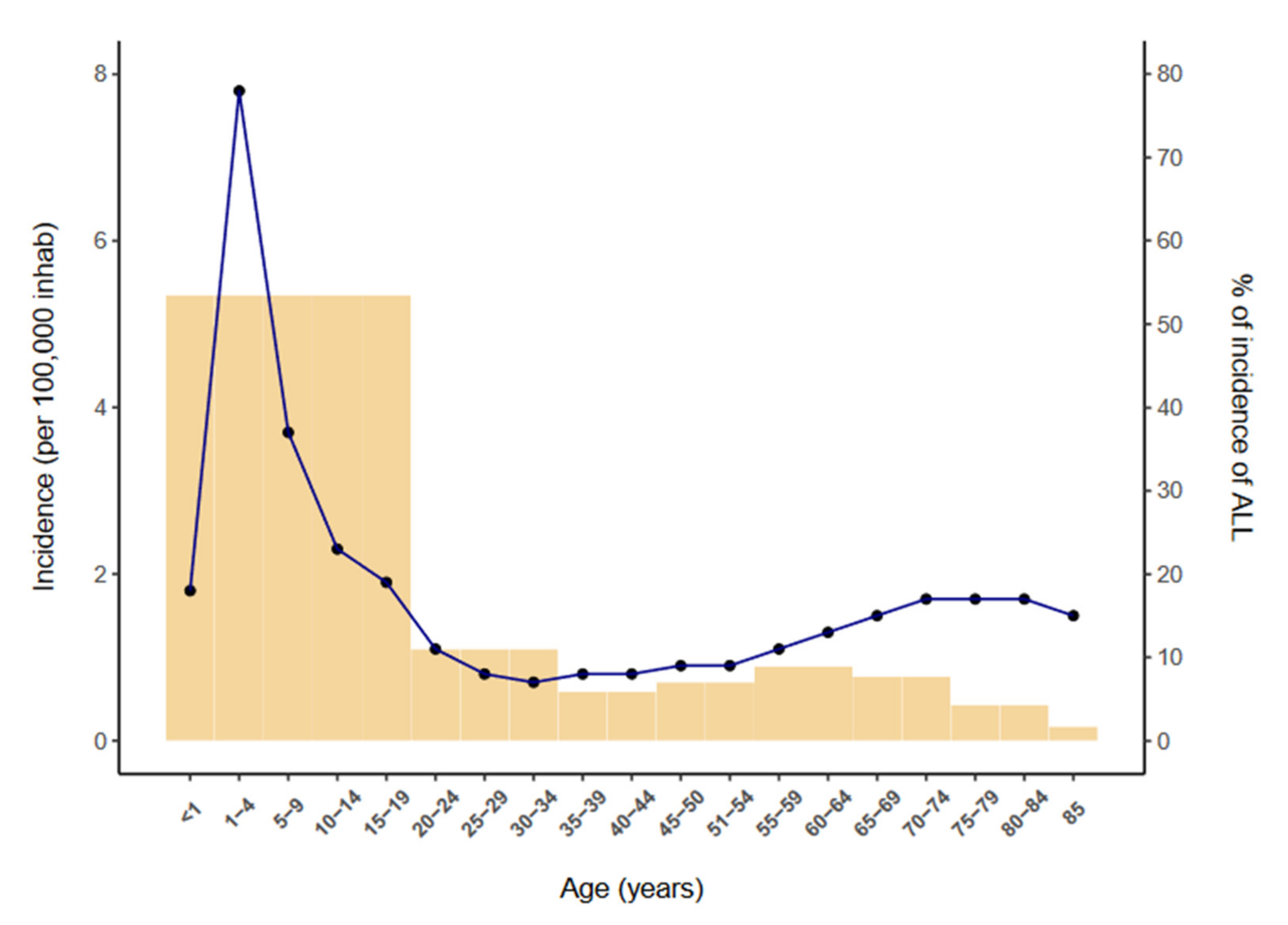
1. Introduction
2. Definition of Hypodiploid B-ALL Subgroups
2.1. Demographic Features of B-ALL with Hypodiploidy <40 Chromosomes
2.2. Clinical and Biological Features of B-ALL with Hypodiploidy <40 Chromosomes
2.3. Extramedullary Involvement at Presentation
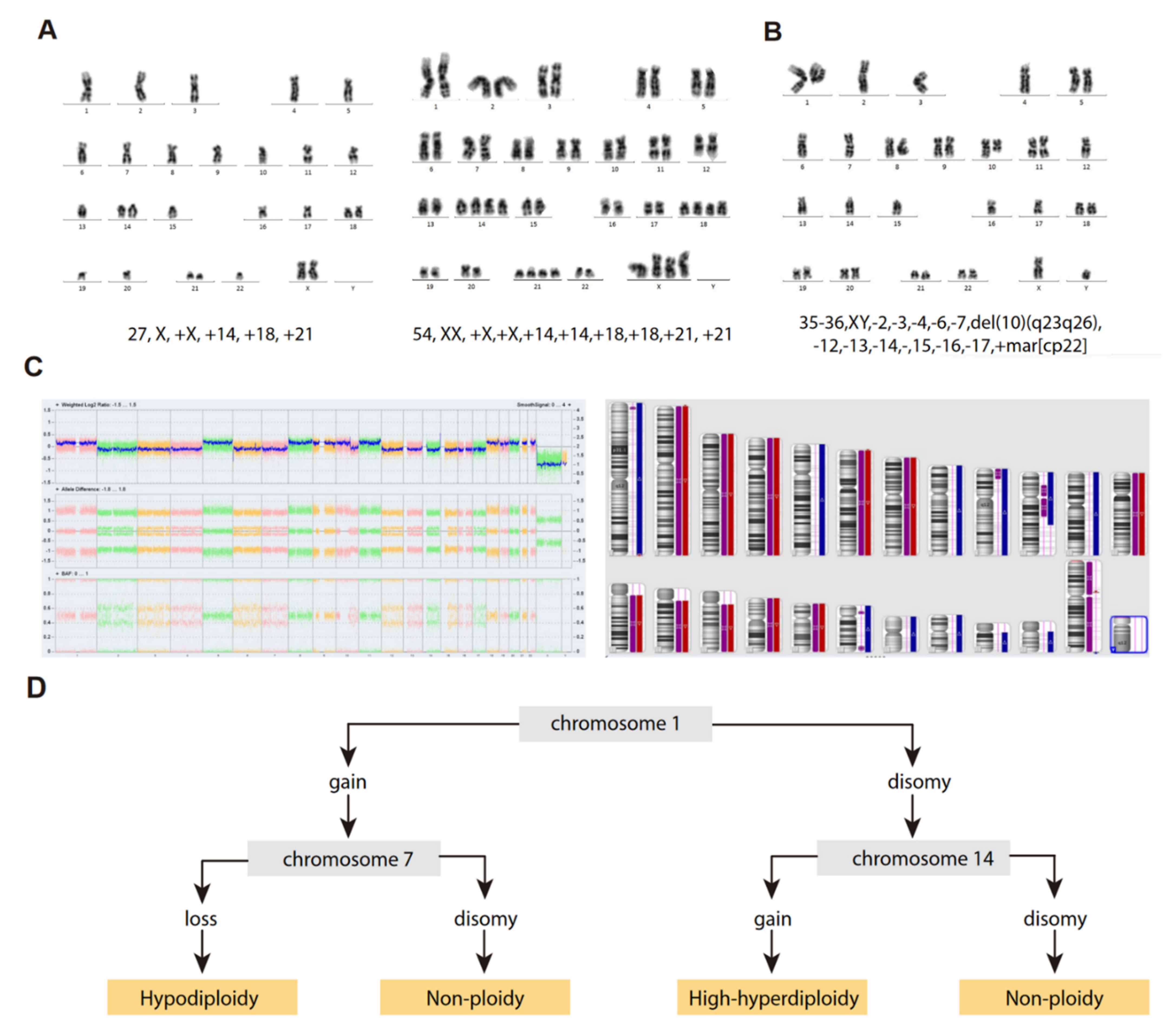
3. Cytogenetic Characterization of B-ALL with Hypodiploidy <40 Chromosomes
“Masked Hypodiploidy”: A Clinical Challenge
4. Molecular Characterization of Hypodiploid B-ALL with <40 Chromosomes
4.1. Near-Haploid B-ALL
4.2. Low-Hypodiploid B-ALL
4.3. Proper Identification of Hypodiploid B-ALL with <40 Chromosomes
5. Etiology of Hypodiploidy in B-ALL
6. Origin of Near-Haploidy and Low-Hypodiploidy
7. Outcome and Treatment Strategies for B-ALL with Hypodiploidies <40 Chromosomes
7.1. Event-Free Survival and Overall Survival Rates
7.2. Relationship of Genetic and Clinical Features with Patient Outcome
7.3. Current Treatment Protocols
7.4. Novel Therapeutic Targets and Approaches to Treat B-ALL with <40 Chromosomes
8. Concluding Remarks
- Both near-haploid and low-hypodiploid B-ALL represent very rare entities, associated with a dismal clinical outcome. Such a low disease incidence represents a challenge to develop pre-clinical models aimed to study the etiology and pathogenesis of the disease and also represents a barrier to design statistically robust clinical trials.
- Cutting-edge cytogenetic and genetic assays must be implemented in routine diagnostic laboratories to distinguish between high-hyperdiploid and masked hypodiploid B-ALL patients since this has a major impact on patient treatment stratification and clinical outcome.
- The pathogenic effect(s) of chromosome losses and its contribution to leukemogenesis is currently not known. Furthermore, the biological contribution of chromosome doublings occurring in most of hypodiploid B-ALL cases with <40 chromosomes is poorly understood. Future studies aiming to decipher the biological mechanisms involved in the progression of hypodiploid B-ALL will help in the identification of innovative targeted treatments and/or diagnostic biomarkers to improve survival in these patients.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.; et al. SEER Cancer Statistics Review (CSR), 1975–2018. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 5 November 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Maitra, A.; McKenna, R.W.; Weinberg, A.G.; Schneider, N.R.; Kroft, S.H. Precursor B-cell lymphoblastic lymphoma. A study of nine cases lacking blood and bone marrow involvement and review of the literature. Am. J. Clin. Pathol. 2001, 115, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulte, D.; Gondos, A.; Brenner, H. Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 2009, 113, 1408–1411. [Google Scholar] [CrossRef]
- Pulte, D.; Jansen, L.; Gondos, A.; Katalinic, A.; Barnes, B.; Ressing, M.; Holleczek, B.; Eberle, A.; Brenner, H.; Group, G.C.S.W. Survival of adults with acute lymphoblastic leukemia in Germany and the United States. PLoS ONE 2014, 9, e85554. [Google Scholar] [CrossRef] [PubMed]
- Kadan-Lottick, N.S.; Ness, K.K.; Bhatia, S.; Gurney, J.G. Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA 2003, 290, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Shuster, J.J.; Wacker, P.; Pullen, J.; Humbert, J.; Land, V.J.; Mahoney, D.H., Jr.; Lauer, S.; Look, A.T.; Borowitz, M.J.; Carroll, A.J.; et al. Prognostic significance of sex in childhood B-precursor acute lymphoblastic leukemia: A Pediatric Oncology Group Study. J. Clin. Oncol. 1998, 16, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Arthur, D.; Camitta, B.; Carroll, A.J.; Crist, W.; Gaynon, P.; Gelber, R.; Heerema, N.; Korn, E.L.; Link, M.; et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J. Clin. Oncol. 1996, 14, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Relling, M.V.; Downing, J.R. Acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.V.; Ensor, H.M.; Richards, S.M.; Chilton, L.; Schwab, C.; Kinsey, S.E.; Vora, A.; Mitchell, C.D.; Harrison, C.J. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: Results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010, 11, 429–438. [Google Scholar] [CrossRef]
- Molina, O.; Abad, M.A.; Sole, F.; Menendez, P. Aneuploidy in Cancer: Lessons from Acute Lymphoblastic Leukemia. Trends Cancer 2021, 7, 37–47. [Google Scholar] [CrossRef]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Bassan, R.; Spinelli, O.; Oldani, E.; Intermesoli, T.; Tosi, M.; Peruta, B.; Rossi, G.; Borlenghi, E.; Pogliani, E.M.; Terruzzi, E.; et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood 2009, 113, 4153–4162. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Williams, D.L.; Raimondi, S.C.; Rivera, G.K.; Look, A.T.; Dodge, R.K.; George, S.L.; Behm, F.G.; Crist, W.M.; Murphy, S.B. Hypodiploidy is associated with a poor prognosis in childhood acute lymphoblastic leukemia. Blood 1987, 70, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.H.; Carroll, A.J.; Raimondi, S.C.; Land, V.J.; Crist, W.M.; Shuster, J.J.; Williams, D.L.; Pullen, D.J.; Borowitz, M.J.; Behm, F.G.; et al. Clinical presentation, karyotypic characterization, and treatment outcome of childhood acute lymphoblastic leukemia with a near-haploid or hypodiploid less than 45 line. Blood 1990, 75, 1170–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chessels, J.M.; Swansbury, G.J.; Reeves, B.; Bailey, C.C.; Richards, S.M. Cytogenetics and prognosis in childhood lymphoblastic leukaemia: Results of MRC UKALL X. Medical Research Council Working Party in Childhood Leukaemia. Br. J. Haematol. 1997, 99, 93–100. [Google Scholar] [CrossRef]
- Heerema, N.A.; Nachman, J.B.; Sather, H.N.; Sensel, M.G.; Lee, M.K.; Hutchinson, R.; Lange, B.J.; Steinherz, P.G.; Bostrom, B.; Gaynon, P.S.; et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: A report from the children’s cancer group. Blood 1999, 94, 4036–4045. [Google Scholar]
- Raimondi, S.C.; Zhou, Y.; Mathew, S.; Shurtleff, S.A.; Sandlund, J.T.; Rivera, G.K.; Behm, F.G.; Pui, C.H. Reassessment of the prognostic significance of hypodiploidy in pediatric patients with acute lymphoblastic leukemia. Cancer 2003, 98, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Moorman, A.V.; Broadfield, Z.J.; Cheung, K.L.; Harris, R.L.; Reza Jalali, G.; Robinson, H.M.; Barber, K.E.; Richards, S.M.; Mitchell, C.D.; et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br. J. Haematol. 2004, 125, 552–559. [Google Scholar] [CrossRef]
- Nachman, J.B.; Heerema, N.A.; Sather, H.; Camitta, B.; Forestier, E.; Harrison, C.J.; Dastugue, N.; Schrappe, M.; Pui, C.H.; Basso, G.; et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 2007, 110, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Jeha, S.; Pei, D.; Payne-Turner, D.; Coustan-Smith, E.; Roberts, K.G.; Waanders, E.; Choi, J.K.; Ma, X.; Raimondi, S.C.; et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 2015, 126, 2896–2899. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cave, H.; Elitzur, S.; Koh, K.; Liu, H.C.; et al. Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef]
- Ishimaru, S.; Okamoto, Y.; Imai, C.; Sakaguchi, H.; Taki, T.; Hasegawa, D.; Cho, Y.; Kakuda, H.; Sano, H.; Manabe, A.; et al. Nationwide survey of pediatric hypodiploid acute lymphoblastic leukemia in Japan. Pediatr. Int. 2019, 61, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- McNeer, J.L.; Devidas, M.; Dai, Y.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; Kahwash, S.B.; Borowitz, M.J.; Wood, B.L.; Larsen, E.; et al. Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Safavi, S.; Paulsson, K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: Two distinct subtypes with consistently poor prognosis. Blood 2017, 129, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, B.; MacCallum, P.; Watts, E.; Rohatiner, A.Z.; Webb, D.; Katz, F.E.; Secker-Walker, L.M.; Temperley, I.J.; Harrison, C.J.; Campbell, R.H.; et al. Near haploid acute lymphoblastic leukemia: Seven new cases and a review of the literature. Leukemia 1991, 5, 738–743. [Google Scholar] [PubMed]
- Moorman, A.V. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012, 26, 123–135. [Google Scholar] [CrossRef]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Groupe Francais de Cytogenetique Hematologique. Cytogenetic abnormalities in adult acute lymphoblastic leukemia: Correlations with hematologic findings outcome. A Collaborative Study of the Group Francais de Cytogenetique Hematologique. Blood 1996, 87, 3135–3142. [Google Scholar]
- Moorman, A.V.; Harrison, C.J.; Buck, G.A.; Richards, S.M.; Secker-Walker, L.M.; Martineau, M.; Vance, G.H.; Cherry, A.M.; Higgins, R.R.; Fielding, A.K.; et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 2007, 109, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Howard, S.C. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet. Oncol. 2008, 9, 257–268. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. An International System for Human Cytogenomic Nomenclature (ISCN 2020); Karger: Basel, Switzerland, 2020; ISBN 978-3-318-06706-4. [Google Scholar]
- Ribera, J.; Granada, I.; Morgades, M.; Vives, S.; Genesca, E.; Gonzalez, C.; Nomdedeu, J.; Escoda, L.; Montesinos, P.; Mercadal, S.; et al. The poor prognosis of low hypodiploidy in adults with B-cell precursor acute lymphoblastic leukaemia is restricted to older adults and elderly patients. Br. J. Haematol. 2019, 186, 263–268. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2021. Available online: https://www.clinicalgenetics.lu.se/division-clinical-genetics/database-chromosome-aberrations-and-gene-fusions-cancer (accessed on 5 November 2021).
- Safavi, S.; Forestier, E.; Golovleva, I.; Barbany, G.; Nord, K.H.; Moorman, A.V.; Harrison, C.J.; Johansson, B.; Paulsson, K. Loss of chromosomes is the primary event in near-haploid and low-hypodiploid acute lymphoblastic leukemia. Leukemia 2013, 27, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Muhlbacher, V.; Zenger, M.; Schnittger, S.; Weissmann, S.; Kunze, F.; Kohlmann, A.; Bellos, F.; Kern, W.; Haferlach, T.; Haferlach, C. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93%. Genes Chromosomes Cancer 2014, 53, 524–536. [Google Scholar] [CrossRef]
- Creasey, T.; Enshaei, A.; Nebral, K.; Schwab, C.; Watts, K.; Cuthbert, G.; Vora, A.; Moppett, J.; Harrison, C.J.; Fielding, A.K.; et al. Single nucleotide polymorphism array-based signature of low hypodiploidy in acute lymphoblastic leukemia. Genes Chromosomes Cancer 2021, 60, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Charrin, C.; Thomas, X.; Ffrench, M.; Le, Q.H.; Andrieux, J.; Mozziconacci, M.J.; Lai, J.L.; Bilhou-Nabera, C.; Michaux, L.; Bernheim, A.; et al. A report from the LALA-94 and LALA-SA groups on hypodiploidy with 30 to 39 chromosomes and near-triploidy: 2 possible expressions of a sole entity conferring poor prognosis in adult acute lymphoblastic leukemia (ALL). Blood 2004, 104, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.J.; Shago, M.; Mikhail, F.M.; Raimondi, S.C.; Hirsch, B.A.; Loh, M.L.; Raetz, E.A.; Borowitz, M.J.; Wood, B.L.; Maloney, K.W.; et al. Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children’s Oncology Group. Cancer Genet. 2019, 238, 62–68. [Google Scholar] [CrossRef]
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.A.N.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood 2021, 138, 948–958. [Google Scholar] [CrossRef]
- Harrison, C.J.; Moorman, A.V.; Barber, K.E.; Broadfield, Z.J.; Cheung, K.L.; Harris, R.L.; Jalali, G.R.; Robinson, H.M.; Strefford, J.C.; Stewart, A.; et al. Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: A UK Cancer Cytogenetics Group Study. Br. J. Haematol. 2005, 129, 520–530. [Google Scholar] [CrossRef]
- Stark, B.; Jeison, M.; Gobuzov, R.; Krug, H.; Glaser-Gabay, L.; Luria, D.; El-Hasid, R.; Harush, M.B.; Avrahami, G.; Fisher, S.; et al. Near haploid childhood acute lymphoblastic leukemia masked by hyperdiploid line: Detection by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2001, 128, 108–113. [Google Scholar] [CrossRef]
- Kohno, S.; Minowada, J.; Sandberg, A.A. Chromosome evolution of near-haploid clones in an established human acute lymphoblastic leukemia cell line (NALM-16). J. Natl. Cancer Inst. 1980, 64, 485–493. [Google Scholar] [PubMed]
- Aburawi, H.E.; Biloglav, A.; Johansson, B.; Paulsson, K. Cytogenetic and molecular genetic characterization of the ‘high hyperdiploid’ B-cell precursor acute lymphoblastic leukaemia cell line MHH-CALL-2 reveals a near-haploid origin. Br. J. Haematol. 2011, 154, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, B.A.; Zielke, N.; Gutierrez, C. Endocycles: A recurrent evolutionary innovation for post-mitotic cell growth. Nat. Rev. Mol. Cell Biol. 2014, 15, 197–210. [Google Scholar] [CrossRef]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safavi, S.; Olsson, L.; Biloglav, A.; Veerla, S.; Blendberg, M.; Tayebwa, J.; Behrendtz, M.; Castor, A.; Hansson, M.; Johansson, B.; et al. Genetic and epigenetic characterization of hypodiploid acute lymphoblastic leukemia. Oncotarget 2015, 6, 42793–42802. [Google Scholar] [CrossRef] [Green Version]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unravelling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Ueno, H.; Yoshida, K.; Shiozawa, Y.; Nannya, Y.; Iijima-Yamashita, Y.; Kiyokawa, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Isobe, T.; et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020, 4, 5165–5173. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stengel, A.; Schnittger, S.; Weissmann, S.; Kuznia, S.; Kern, W.; Kohlmann, A.; Haferlach, T.; Haferlach, C. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood 2014, 124, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.V. Does TP53 guard ALL genomes? Blood 2014, 124, 160–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.; Kobayashi, S.; Pauly, M.; Lew, G.; Saxe, D.; Keller, F.; Qayed, M.; Castellino, S. Evaluating approaches to enhance survival in children with hypodiploid acute lymphoblastic leukaemia (ALL). Br. J. Haematol. 2019, 185, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Comeaux, E.Q.; Mullighan, C.G. TP53 Mutations in Hypodiploid Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026286. [Google Scholar] [CrossRef] [Green Version]
- Lafage-Pochitaloff, M.; Baranger, L.; Hunault, M.; Cuccuini, W.; Lefebvre, C.; Bidet, A.; Tigaud, I.; Eclache, V.; Delabesse, E.; Bilhou-Nabera, C.; et al. Impact of cytogenetic abnormalities in adults with Ph-negative B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1832–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsson, K.; Lilljebjorn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjogren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676. [Google Scholar] [CrossRef]
- Paulsson, K. High hyperdiploid childhood acute lymphoblastic leukemia: Chromosomal gains as the main driver event. Mol. Cell. Oncol. 2016, 3, e1064555. [Google Scholar] [CrossRef] [Green Version]
- Mandahl, N.; Johansson, B.; Mertens, F.; Mitelman, F. Disease-associated patterns of disomic chromosomes in hyperhaploid neoplasms. Genes Chromosomes Cancer 2012, 51, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Molina, O. (Josep Carreras Leukemia Research Institute, Barcelona, Spain). High-resolution confocal microscopy analyses of mitotic progression using PDX samples derived from near-haploid and low-hypodiploid B-ALL. 2021; material not intended for publication.
- Gruhn, B.; Taub, J.W.; Ge, Y.; Beck, J.F.; Zell, R.; Hafer, R.; Hermann, F.H.; Debatin, K.M.; Steinbach, D. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia 2008, 22, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 471–484. [Google Scholar] [CrossRef]
- Schultz, K.R.; Devidas, M.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Zheng, H.W.; Davies, S.M.; Gaynon, P.S.; Trigg, M.; et al. Philadelphia chromosome-negative very high-risk acute lymphoblastic leukemia in children and adolescents: Results from Children’s Oncology Group Study AALL0031. Leukemia 2014, 28, 964–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.A.; Zhang, M.J.; Eapen, M.; He, W.; Seber, A.; Gibson, B.; Camitta, B.M.; Kitko, C.L.; Dvorak, C.C.; Nemecek, E.R.; et al. Transplantation Outcomes for Children with Hypodiploid Acute Lymphoblastic Leukemia. Biol. Blood Marrow Transplant. 2015, 21, 1273–1277. [Google Scholar] [CrossRef] [Green Version]
- Groeneveld-Krentz, S.; Schroeder, M.P.; Reiter, M.; Pogodzinski, M.J.; Pimentel-Gutierrez, H.J.; Vagkopoulou, R.; Hof, J.; Chen-Santel, C.; Nebral, K.; Bradtke, J.; et al. Aneuploidy in children with relapsed B-cell precursor acute lymphoblastic leukaemia: Clinical importance of detecting a hypodiploid origin of relapse. Br. J. Haematol. 2019, 185, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Amare, P.K.; Mohanty, P.; Talker, E.; Chaubal, K.; Jain, H.; Tembhare, P.; Patkar, N.; Chaturvedi, A.; Subramanian, P.G.; et al. Investigating the clinical, hematological and cytogenetic profile of endoreduplicated hypodiploids in BCP-ALL. Blood Cells Mol. Dis. 2020, 85, 102465. [Google Scholar] [CrossRef]
- Winter, G.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; Escherich, G.; Moricke, A.; von Stackelberg, A.; Stanulla, M.; Bailey, S.; Richter, L.; Steinemann, D.; et al. Clinical and genetic characteristics of children with acute lymphoblastic leukemia and Li-Fraumeni syndrome. Leukemia 2021, 35, 1475–1479. [Google Scholar] [CrossRef]
- Lee, S.H.R.; Li, Z.; Tai, S.T.; Oh, B.L.Z.; Yeoh, A.E.J. Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers 2021, 13, 4068. [Google Scholar] [CrossRef] [PubMed]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef]
- Talleur, A.C.; Maude, S.L. What is the role for HSCT or immunotherapy in pediatric hypodiploid B-cell acute lymphoblastic leukemia? Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 508–511. [Google Scholar] [CrossRef]
- Ribera, J.M.; Oriol, A.; Morgades, M.; Montesinos, P.; Sarra, J.; Gonzalez-Campos, J.; Brunet, S.; Tormo, M.; Fernandez-Abellan, P.; Guardia, R.; et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: Final results of the PETHEMA ALL-AR-03 trial. J. Clin. Oncol. 2014, 32, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, E.; Comeaux, E.Q.; Kim, K.L.; Melnik, E.; Beckman, K.; Davis, K.L.; Wu, K.; Akutagawa, J.; Bridges, O.; Marino, R.; et al. Bcl-2 Is a Therapeutic Target for Hypodiploid B-Lineage Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 2339–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Becker, P.S.; Linenberger, M.; Eaton, K.D.; Appelbaum, F.R.; Dreyer, Z.; Airewele, G.; Redell, M.; Lopez-Terrada, D.; Patel, A.; et al. Adult Low-Hypodiploid Acute B-Lymphoblastic Leukemia With IKZF3 Deletion and TP53 Mutation: Comparison With Pediatric Patients. Am. J. Clin. Pathol. 2015, 144, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Diorio, C.; Maude, S.L. CAR T cells vs allogeneic HSCT for poor-risk ALL. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 501–507. [Google Scholar] [CrossRef]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood 2021, 137, 471–484. [Google Scholar] [CrossRef]
- Wang, L.; Ashraf, D.C.; Kinde, B.; Ohgami, R.S.; Kumar, J.; Kersten, R.C. Hypodiploid B-Lymphoblastic Leukemia Presenting as an Isolated Orbital Mass Prior to Systemic Involvement: A Case Report and Review of the Literature. Diagnostics 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Sposto, R.; Shah, N.N.; Rodriguez, V.; Yuan, C.; Stetler-Stevenson, M.; O’Brien, M.M.; McNeer, J.L.; Quereshi, A.; Cabannes, A.; et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 2019, 33, 884–892. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Precursor Lymphoid Neoplasms |
|---|
| B-lymphoblastic leukemia/lymphoma not otherwise specified (NOS) |
B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities
|
T-lymphoblastic leukemia/lymphoma
|
| NK-lymphoblastic leukemia/lymphoma |
| Study | Near-Haploid | Low-Hypodiploid | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M/F 1 | Age | WBC (×109/L) * | PreB/T Lineage | M/F 1 | Age | WBC (×109/L) * | PreB/T Lineage | |||||||||
| M 2 | <10 yo at dx * | M 2 | ≤20 | 20–50 | >50 | M 2 | <10 yo at dx * | M 2 | ≤20 | 20–50 | >50 | |||||
| SJCRH [16] | nr | 4.5 | 3 | nr | nr | nr | 11 | 1 | nr | nr | ||||||
| SJCRH-POG [17] | 0.66 | 4.7 | 70 | 13 | 70 | 10 | 20 | 8/13 | 2 | 10.5 | 22.2 | 8.4 | 66.6 | 11.1 | 22.2 | 5/1 3 |
| CCG [19] | 63 | 50 | 13 | 38 | nr | nr | nr | nr | ||||||||
| SJCRH [20] | 7.3 | 100 | 2.5 | 100 | 0 | 0 | 4/0 | nr | 0 | 8.2 | 83.3 | 0 | 16.6 | 5/1 | ||
| MRC [21] | 0.75 | 7.4 | 64.3 | 67.3 | 38.5 | 30.8 | 30.8 | 13/0 3 | 1.1 | 21.6 | 7.1 | 11 | 85.7 | 7.1 | 7.1 | 12/0 3,4 |
| Inc study [22] | 1 | 69.6 | 39.1 | 41.3 | 19.6 | nr | 2.25 | 19.2 | 73 | 19.2 | 7.7 | nr | ||||
| SJTTS 15&16 [23] | 3.6 | 8.1 | 13.9 | 5.6 | ||||||||||||
| PDLWG [24] | 1.46 | 6.2 | 73 | 21.8 | 49 | 20 | 31 | 99/1 | 1.14 | 12.9 | 26 | 7 | 85 | 10 | 4 | 99/1 |
| Japanese study [25] | nr | nr | nr | nr | nr | nr | nr | 3/0 | nr | nr | nr | nr | nr | nr | nr | 3/0 |
| Age (Years) | Near-Haploid | Low-Hypodiploid | Reference | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MN | Retained chr | Lost chr | Doubled Clone | Frequency | MN | Retained chr | Lost chr | Doubled Clone | Frequency | ||
| 1–18 | 25–28 | 8, 10, 14, 18, 21 and sex chr. | yes | 0.0046 | 30–40 | 1, 19, 21, 22 and sex chr. | 3, 7, 13, 16, 17 | yes | 0.41 | [17] | |
| 1–10 | 24–28 | 8, 10, 14, 18, 21 and sex chr. | 7, 13, 14, 20, X | yes | 0.0042 | 33–44 | 7, 13, 14, 20, X | nr | 0.79 | [19] | |
| 2–15 | 23–29 | 14, 18, 21 and sex chr. | yes | 0.0039 | 33–39 | 1, 2, 5, 6, 8, 10, 11,12, 14, 18, 19, 21, 22 and the sex chr. | 7, 17 | yes | 0.39 | [21] | |
| 15–84 | - | - | - | - | - | 30–39 | 1, 5, 6, 8, 10, 11, 15, 18, 19, 21, 22, X, Y | 3, 7, 15, 16, 17 | 66 to 78 chr | 0.05 | [31] |
| 15–55 | - | - | - | - | - | 33–39 | nr | nr | nr | 0.0008 | [21] |
| 15–55/>55 | <30 | 0.0016 | 32–39 | 1 | 2, 3, 7, 9, 13, 15, 16, 17, 20, 4 | 64–74 | 3.85% | [40] | |||
| 1–9/>10 | 24–29 | 14, 18, 21 and sex chr. | nr | nr | nr | 33–39 | nrec | 3, 7, 16, 17 | nr | nr | [10] |
| <31 | 24–31 | 14, 18, 21 and sex chr. | nr | yes | 0.008 | 32–39 | 1, 8, 10, 11, 18, 19, 21 and 22 | nr | nr | 0.0064 | [41] |
| Genes | Cellular Pathway | Near-Haploid B-ALL | Low-Hypodiploid B-ALL | ||||
|---|---|---|---|---|---|---|---|
| Mutation | Focal Deletion | Focal DEL + Mut | Mutation | Focal Deletion | Focal DEL + Mut | ||
| NF1 | RTK/RAS pathway | 11/68 (16%) | 16/68 (24%) | 3/68 (4%) | 0 | 2/34 (6%) | 0 |
| KRAS | 2/68 (3%) | 0 | 0 | 0 | 0 | 0 | |
| NRAS | 10/68 (15%) | 0 | 0 | 0 | 0 | 0 | |
| PTPN11 | 1/68 (1%) | 0 | 0 | 0 | 0 | 0 | |
| FLT3 | 6/68 (9%) | 0 | 0 | 0 | 0 | 0 | |
| CRLF2 | 0 | 2/68 (3%) * | 0 | 0 | 0 | 0 | |
| MAPK1 | 1/68 (1%) | 0 | 0 | 0 | 0 | 0 | |
| GAB2 | 0 | 2/68 (3%) | 0 | 0 | 1/34 (3%) | 0 | |
| EPHA7 | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 | |
| RASA2 | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 | |
| IKZF1 | B-cell development | 0 | 3/68 (4%) | 0 | 0 | 1/34 (3%) | 0 |
| IKZF2 | 1/68 (1%) | 0 | 0 | 0 | 18/34 (53%) | 0 | |
| IKZF3 | 1/68 (1%) | 8/68 (12%) | 0 | 0 | 1/34 (3%) | 0 | |
| PAX5 | 1/68 (1%) | 4/68 (6%) | 0 | 0 | 2/34 (6%) | 0 | |
| EBF1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| VPREB1 | 0 | 3/68 (4%) | 0 | 0 | 2/34 (6%) | 0 | |
| CDKN2A/B | Cell cycle and apoptosis | 0 | 15/68 (22%) | 0 | 0 | 8/34 (24%) | 0 |
| TP53 | 2/68 (3%) | 0 | 0 | 31/34 (91%) | 0 | 0 | |
| RB1 | 2/68 (3%) | 3/68 (4%) | 1/68 (1%) | 5/34 (15%) | 8/34 (24%) | 0 | |
| ETV6 | Hematopoiesis | 1/68 (1%) | 3/68 (4%) | 1/68 (1%) | 0 | 0 | 0 |
| Histone cluster (6p22) | Histone-related | 0 | 13/68 (19%) | 0 | 0 | 1/34 (3%) | 0 |
| ARID1B | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 | |
| PAG1 | BCR signalling | 1/68 (1%) | 6/68 (9%) | 0 | 0 | 1/34 (3%) | 0 |
| ARPP21 | Calmodulin signalling | 0 | 1/68 (1%) | 0 | 0 | 0 | 0 |
| SLX4IP (C20orf194) | Telomere length maintenance | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 |
| CUL5 | Ubiquitin pathway | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 |
| FAM53B | Wnt signalling | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 |
| PDS5B (APRIN) | Cohesin complex | 0 | 2/68 (3%) | 0 | 0 | 0 | 0 |
| ANKRD11 | Cell adhesion | 0 | 0 | 0 | 0 | 2/34 (6%) | 0 |
| DMD | 0 | 0 | 0 | 0 | 1/34 (3%) | 0 | |
| Study group | Years | Outcome variable | MN | Other variables | n | Mean (%) | p-value | Reference |
|---|---|---|---|---|---|---|---|---|
| Total Therapy Studies IX–XI Pediatric Oncology Group (POG) | 1979–1988 1986-1988 | 3-year-DFS | <30 chr 30–40 chr 41–44 chr | 109 8 | 40 33 37 | [17] | ||
| Children’s Cancer Group (CCG) | 1988–1995 | 6-year-EFS | 33–34 chr 29–32 chr 24–28 chr | 15 | 40 | [19] | ||
| 30–40 chr | 0 | NA | ||||||
| 41–44 chr | 8 | 25 | ||||||
| Total Therapy protocols T11–T14 | 1984–1999 | 5-years EFS | 36–44 chr | 17 | [20] | |||
| 25–29 chr | 26 | |||||||
| ≥45 chr | 75 | <0.001 | ||||||
| <45 chr | 20 | |||||||
| Medical Research Council, UK ALL trial protocols | 1990–2002 | 3-year-EFS | 42–45 chr | 121 | 65 | 0.0002 | [21] | |
| 25–39 chr | 20 | 29 | ||||||
| AIEOP-3; BFM-5; CCG-33; COALL-3; DANA FARBER-4; POG-44; SJCRH-6; UK-20; NOPHO-6; and EORTC-15 | 1986–1996 | 8-year EFS | 44 chr | 50 | 52 | <0.01 | [22] | |
| 40–43 chr | 8 | 19 | ||||||
| 33–39 chr | 26 | 37 | ||||||
| 30–32 chr | 0 | NA | ||||||
| 24–29 chr | 46 | 28 | ||||||
| COG AALL0031 | 2002–2006 | 4-year EFS | <30–44 chr | Chemotherapy alone | 26 | 50 | 0.65 | [66] |
| Chemotherapy + BMT | 13 | 62 | ||||||
| St. Jude Total Therapy Study XV&XVI | 2000–2014 | 5-year EFS | 24–31 chr | 8 | 73 | 0.8 | [23] | |
| 32–39 chr | 12 | 75 | ||||||
| MRD EOI neg | 14 | 85 | 0.03 | |||||
| MRD EOI pos | 6 | 44 | ||||||
| CIBMTR-All BMT in CR1 or CR2 | 1990–2010 | 5-year-DFS | 44–45 chr | 39 | 64 | 0.01 | [67] | |
| ≤43 chr | 39 | 37 | ||||||
| Children’s Healthcare of Atlanta | 2004–2016 | 2-year DFS | 32–39 chr | 5 | 60 | 0.853 | [57] | |
| 24–31 chr | 7 | 71 | ||||||
| MRD EOI neg | 10 | 69 | ||||||
| MRD EOI pos | 2 | 50 | ||||||
| Age ≥ 10 years | 6 | 33 | 0.021 | |||||
| Age < 10 years | 6 | 100 | ||||||
| COG AALL03B1-COG AALL0331 and AALL0232 * | 2003–2011 | 5-year EFS | >46 chr | NR | 85 | <0.01 | [26] | |
| <44 chr | 131 | 52 | ||||||
| BMT en CR1 | 61 | 56 | 0.62 | |||||
| No BMT | 52 | 49 | ||||||
| MRD pos | 30 | 26 | ||||||
| MRD neg | 74 | 64 | ||||||
| Standard NCI risk group | 48 | 60 | 0.026 | |||||
| High NCI risk group | 83 | 47 | ||||||
| Chemotherapy 1 | 4 | 0.13 | ||||||
| HSCT 1 | 7 | |||||||
| Ponte di Legno Childhood ALL Working Group- ≤44 chromosomas | 1997–2013 | 5-year EFS | 44 chr | 40 | 74 | 0.053 | [24] | |
| 40–43 chr | 13 | 58 | ||||||
| 30–39 chr | 118 | 50 | ||||||
| 24–29 chr | 101 | 56 | ||||||
| HCT in LH | 21 | 64 | 0.89 | |||||
| No HCT in LH | 93 | 62 | ||||||
| HCT in NH | 19 | 51 | 0.6 | |||||
| No HCT in NH | 82 | 44 | ||||||
| TCCSG, JACLS, Japanese Children’s Cancer and Leukemia Study Group, Kyushu-Yamaguchi Children’s Cancer Study Group | 1997–2012 | 5-year EFS | 45 chr | 101 | 73 | <0.036 | [25] | |
| 44 chr | 8 | 88 | ||||||
| <44 chr | 8 | 38 | ||||||
| Relapse rate in patients < 44 chr | 5 | 63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molina, O.; Bataller, A.; Thampi, N.; Ribera, J.; Granada, I.; Velasco, P.; Fuster, J.L.; Menéndez, P. Near-Haploidy and Low-Hypodiploidy in B-Cell Acute Lymphoblastic Leukemia: When Less Is Too Much. Cancers 2022, 14, 32. https://doi.org/10.3390/cancers14010032
Molina O, Bataller A, Thampi N, Ribera J, Granada I, Velasco P, Fuster JL, Menéndez P. Near-Haploidy and Low-Hypodiploidy in B-Cell Acute Lymphoblastic Leukemia: When Less Is Too Much. Cancers. 2022; 14(1):32. https://doi.org/10.3390/cancers14010032
Chicago/Turabian StyleMolina, Oscar, Alex Bataller, Namitha Thampi, Jordi Ribera, Isabel Granada, Pablo Velasco, José Luis Fuster, and Pablo Menéndez. 2022. "Near-Haploidy and Low-Hypodiploidy in B-Cell Acute Lymphoblastic Leukemia: When Less Is Too Much" Cancers 14, no. 1: 32. https://doi.org/10.3390/cancers14010032
APA StyleMolina, O., Bataller, A., Thampi, N., Ribera, J., Granada, I., Velasco, P., Fuster, J. L., & Menéndez, P. (2022). Near-Haploidy and Low-Hypodiploidy in B-Cell Acute Lymphoblastic Leukemia: When Less Is Too Much. Cancers, 14(1), 32. https://doi.org/10.3390/cancers14010032