
Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes

 ,
,  , , ,
, , ,  ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
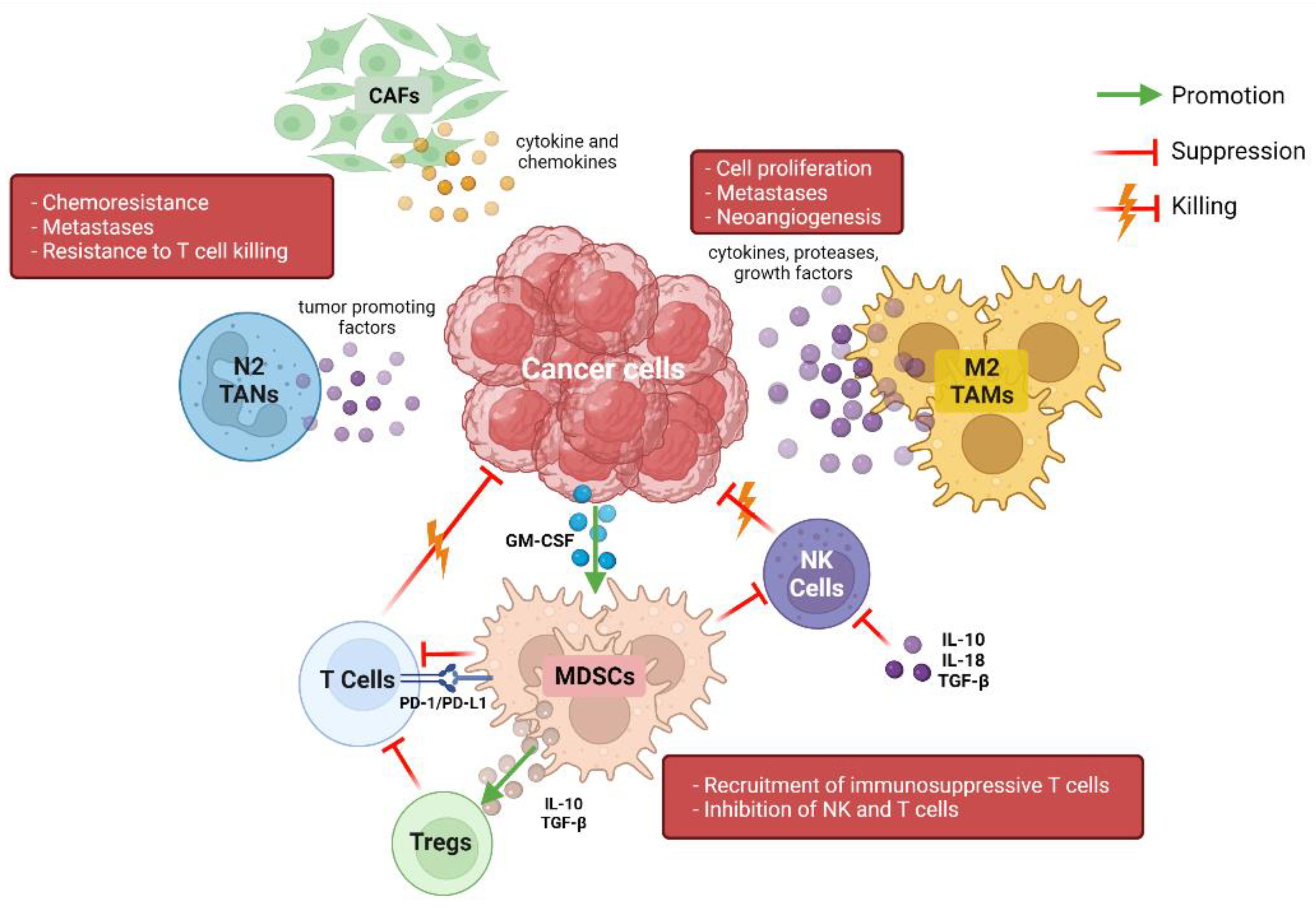
2. The Pancreatic Tumor Immune Microenvironment
3. Prognostic and Predictive Immune Biomarkers
Immunotherapy: Searching for the Right Key
4. Conclusions
Funding
Conflicts of Interest
References
- National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Pancreatic Cancer. 2022. Available online: https://seer.cancer.gov/statfacts/html/pancreas (accessed on 5 March 2022).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, M.; Wu, Z.; Krautz, C.; Pilarsky, C.; Grützmann, R.; Weber, G.F. Current Clinical Strategies of Pancreatic Cancer Treatment and Open Molecular Questions. Int. J. Mol. Sci. 2019, 20, 4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint Inhibitors in Pancreatic Cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Farrow, B.; Albo, D.; Berger, D.H. The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and Macrophage Heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Di Caro, G.; Cortese, N.; Castino, G.F.; Grizzi, F.; Gavazzi, F.; Ridolfi, C.; Capretti, G.; Mineri, R.; Todoric, J.; Zerbi, A.; et al. Dual Prognostic Significance of Tumour-Associated Macrophages in Human Pancreatic Adenocarcinoma Treated or Untreated with Chemotherapy. Gut 2016, 65, 1710–1720. [Google Scholar] [CrossRef]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological Macrophage Inhibition Decreases Metastasis Formation in a Genetic Model of Pancreatic Cancer. Gut 2017, 66, 1278–1285. [Google Scholar] [CrossRef]
- Filippini, D.; Agosto, S.D.; Delfino, P.; Simbolo, M.; Piro, G.; Rusev, B.; Veghini, L.; Cantù, C.; Lupo, F.; Ugel, S.; et al. Immunoevolution of Mouse Pancreatic Organoid Isografts from Preinvasive to Metastatic Disease. Sci. Rep. 2019, 9, 12286. [Google Scholar] [CrossRef] [Green Version]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages Mediate Gemcitabine Resistance of Pancreatic Adenocarcinoma by Upregulating Cytidine Deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage Plasticity and Interaction with Lymphocyte Subsets: Cancer as a Paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-Talk between Myeloid-Derived Suppressor Cells (MDSC), Macrophages, and Dendritic Cells Enhances Tumor-Induced Immune Suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T Cells Sustain Myeloid-Derived Suppressor Cell-Mediated Immune Suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Dhar, P.; Wu, J.D. NK Cell Plasticity in Cancer. J. Clin. Med. 2019, 8, 1492. [Google Scholar] [CrossRef] [Green Version]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56(Bright)Perforin(Low) Noncytotoxic Human NK Cells Are Abundant in Both Healthy and Neoplastic Solid Tissues and Recirculate to Secondary Lymphoid Organs via Afferent Lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, S.M.; Wen, J.; Moutsopoulos, N.M. The Kiss of Death: Interrupted by NK-Cell Close Encounters of Another Kind. Trends Immunol. 2006, 27, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor-Beta-Dependent Manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Funa, K.; Nilsson, B.; Jacobsson, G.; Alm, G.V. Decreased Natural Killer Cell Activity and Interferon Production by Leucocytes in Patients with Adenocarcinoma of the Pancreas. Br. J. Cancer 1984, 50, 231–233. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.W.; et al. Defective Localization with Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Hirth, M.; Gandla, J.; Höper, C.; Gaida, M.M.; Agarwal, N.; Simonetti, M.; Demir, A.; Xie, Y.; Weiss, C.; Michalski, C.W.; et al. CXCL10 and CCL21 Promote Migration of Pancreatic Cancer Cells Toward Sensory Neurons and Neural Remodeling in Tumors in Mice, Associated with Pain in Patients. Gastroenterology 2020, 159, 665–681.e13. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Xiang, Z.J.; Hu, T.; Wang, Y.; Wang, H.; Xu, L.; Cui, N. Neutrophil-Lymphocyte Ratio (NLR) Was Associated with Prognosis and Immunomodulatory in Patients with Pancreatic Ductal Adenocarcinoma (PDAC). Biosci. Rep. 2020, 40, BSR20201190. [Google Scholar] [CrossRef]
- Brandau, S.; Dumitru, C.A.; Lang, S. Protumor and Antitumor Functions of Neutrophil Granulocytes. Semin. Immunopathol. 2013, 35, 163–176. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandra, V.; Sanchez, E.R.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-Induced Neutrophil Extracellular Traps Mediate Resistance to Checkpoint Blockade in Pancreatic Cancer. J. Exp. Med. 2020, 217, 20190354. [Google Scholar] [CrossRef]
- Tao, L.; Zhang, L.; Peng, Y.; Tao, M.; Li, L.; Xiu, D.; Yuan, C.; Ma, Z.; Jiang, B. Neutrophils Assist the Metastasis of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma: A New Hypothesis and a New Predictor for Distant Metastasis. Medicine 2016, 95, e4932. [Google Scholar] [CrossRef] [PubMed]
- Nasca, V.; Chiaravalli, M.; Piro, G.; Esposito, A.; Salvatore, L.; Tortora, G.; Corbo, V.; Carbone, C. Intraductal Pancreatic Mucinous Neoplasms: A Tumor-Biology Based Approach for Risk Stratification. Int. J. Mol. Sci. 2020, 21, 6386. [Google Scholar] [CrossRef] [PubMed]
- Vizio, B.; Novarino, A.; Giacobino, A.; Cristiano, C.; Prati, A.; Ciuffreda, L.; Montrucchio, G.; Bellone, G. Potential Plasticity of T Regulatory Cells in Pancreatic Carcinoma in Relation to Disease Progression and Outcome. Exp. Ther. Med. 2012, 4, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Kleinewietfeld, M.; Hafler, D.A. The Plasticity of Human Treg and Th17 Cells and Its Role in Autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, M.J. Expanding Roles for CD4 T Cells and Their Subpopulations in Tumor Immunity and Therapy. Front. Oncol. 2013, 3, 63. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T Helper Type 2 Cell Infiltrate Correlates with Cancer-Associated Fibroblast Thymic Stromal Lymphopoietin Production and Reduced Survival in Pancreatic Cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- Tassi, E.; Gavazzi, F.; Albarello, L.; Senyukov, V.; Longhi, R.; Dellabona, P.; Doglioni, C.; Braga, M.; Di Carlo, V.; Protti, M.P. Carcinoembryonic Antigen-Specific but Not Antiviral CD4+ T Cell Immunity Is Impaired in Pancreatic Carcinoma Patients. J. Immunol. 2008, 181, 6595–6603. [Google Scholar] [CrossRef] [Green Version]
- Piro, G.; Simionato, F.; Carbone, C.; Frizziero, M.; Malleo, G.; Zanini, S.; Casolino, R.; Santoro, R.; Mina, M.M.; Zecchetto, C.; et al. A Circulating T H 2 Cytokines Profile Predicts Survival in Patients with Resectable Pancreatic Adenocarcinoma. Oncoimmunology 2017, 6, e1322242. [Google Scholar] [CrossRef] [Green Version]
- Prokopchuk, O.; Liu, Y.; Henne-Bruns, D.; Kornmann, M. Interleukin-4 Enhances Proliferation of Human Pancreatic Cancer Cells: Evidence for Autocrine and Paracrine Actions. Br. J. Cancer 2005, 92, 921–928. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Wörmann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truit, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Liu, X.; Liang, C.; Hua, J.; Xu, J.; Wang, W.; Meng, Q.; Liu, J.; Zhang, B.; Yu, X.; et al. Deciphering the Prognostic Implications of the Components and Signatures in the Immune Microenvironment of Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2021, 12, 648917. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal Biology and Therapy in Pancreatic Cancer: Ready for Clinical Translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Belle, J.I.; Denardo, D.G. A Single-Cell Window into Pancreas Cancer Fibroblast Heterogeneity. Cancer Discov. 2019, 9, 1001–1002. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b Study of a Small Molecule Antagonist of Human Chemokine (C-C Motif) Receptor 2 (PF-04136309) in Combination with Nab-Paclitaxel/Gemcitabine in First-Line Treatment of Metastatic Pancreatic Ductal Adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.G.; Shon, Y.; Kim, J.; Oh, Y.K. Selective Activation of Anticancer Chemotherapy by Cancer-Associated Fibroblasts in the Tumor Microenvironment. J. Natl. Cancer Inst. 2016, 109, djw186. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF Mediates Proinvasive Activation of Stromal Fibroblasts in Cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus Gemcitabine vs. Gemcitabine for First-Line Treatment of Patients with Unresectable Pancreatic Cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Madden, J. Infinity reports update from Phase 2 study of Saridegib plus Gemcitabine in patients with metastatic pancreatic cancer. Infin. Pharm. 2012, 2012–2014. [Google Scholar]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor Mutational Load Predicts Survival after Immunotherapy across Multiple Cancer Types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Gibbs, P. Inflammation, Biomarkers and Immuno-Oncology Pathways in Pancreatic Cancer. J. Pers. Med. 2019, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, R.T.; Mattiolo, P.; Mafficini, A.; Hong, S.M.; Piredda, M.L.; Taormina, S.V.; Malleo, G.; Marchegiani, G.; Pea, A.; Salvia, R.; et al. Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Pancreatic Cancer: Systematic Review and Still-Open Questions. Cancers 2021, 13, 3119. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Karamitopoulou, E.; Andreou, A.; de Mortanges, A.P.; Tinguely, M.; Gloor, B.; Perren, A. PD-1/PD-L1-Associated Immunoarchitectural Patterns Stratify Pancreatic Cancer Patients into Prognostic/Predictive Subgroups. Cancer Immunol. Res. 2021, 9, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.J.; Coleman, H.G.; McCain, R.S.; Kelly, P.J.; Johnston, D.I.; Taylor, M.A.; Turkington, R.C. Immune Cell Infiltrates as Prognostic Biomarkers in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis. J. Pathol. Clin. Res. 2021, 7, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Wei, X.; Jiang, H.; Lan, C.; Yang, S.; Wang, H.; Yang, Y.; Tian, C.; Xu, Z.; et al. PD-L1 Is a Direct Target of Cancer-FOXP3 in Pancreatic Ductal Adenocarcinoma (PDAC), and Combined Immunotherapy with Antibodies against PD-L1 and CCL5 Is Effective in the Treatment of PDAC. Signal Transduct. Target. Ther. 2020, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, M.A.; de Carné Trécesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 MRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC Regulates the Antitumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J. Natl. Cancer Inst. 2017, 109, djw283. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Macherla, S.; Laks, S.; Naqash, A.R.; Bulumulle, A.; Zervos, E.; Muzaffar, M. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive Characterisation of Pancreatic Ductal Adenocarcinoma with Microsatellite Instability: Histology, Molecular Pathology and Clinical Implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.D.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- El-Jawhari, J.J.; El-Sherbiny, Y.M.; Scott, G.B.; Morgan, R.S.M.; Prestwich, R.; Bowles, P.A.; Blair, G.E.; Tanaka, T.; Rabbitts, T.H.; Meade, J.L.; et al. Blocking Oncogenic RAS Enhances Tumour Cell Surface MHC Class I Expression but Does Not Alter Susceptibility to Cytotoxic Lymphocytes. Mol. Immunol. 2014, 58, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Hester, R.; Mazur, P.K.; McAllister, F. Immunotherapy in Pancreatic Adenocarcinoma: Beyond “Copy/Paste”. Clin. Cancer Res. 2021, 27, 6287–6297. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of Immune Infiltrates in Melanoma and Pancreatic Cancer Highlights VISTA as a Potential Target in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Liu, Z.; Rangelova, E.; Poiret, T.; Ambati, A.; Rane, L.; Xie, S.; Verbeke, C.; Dodoo, E.; Del Chiaro, M.; et al. Expansion of Tumor-Reactive T Cells from Patients with Pancreatic Cancer. J. Immunother. 2016, 39, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Manero-Rupérez, N.; Martínez-Bosch, N.; Barranco, L.E.; Visa, L.; Navarro, P. The Galectin Family as Molecular Targets: Hopes for Defeating Pancreatic Cancer. Cells 2020, 9, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The Interaction of TIGIT with PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8 + T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Byrne, K.T.; Vonderheide, R.H. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef] [Green Version]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.L.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-Cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, with or without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II Study of Gemcitabine, Nab-Paclitaxel, and Pembrolizumab in Metastatic Pancreatic Adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Knox, J.J.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; Harb, M.; Aucoin, N.; et al. LBA65 The Canadian Cancer Trials Group PA.7 Trial: Results of a Randomized Phase II Study of Gemcitabine (GEM) and Nab-Paclitaxel (Nab-P) vs GEM, Nab-P, Durvalumab (D) and Tremelimumab (T) as First Line Therapy in Metastatic Pancreatic Ductal Adenocarcinoma (MPDAC). Ann. Oncol. 2020, 31, S1195. [Google Scholar] [CrossRef]
- Rahma, O.E.; Katz, M.H.G.; Wolpin, B.M.; Dias-Costa, A.; Nowak, J.; Rodig, S.J.; Dougan, S.; Bekaii-Saab, T.S.; Stucky, C.-C.H.; Elias, R.; et al. Randomized Multicenter Phase Ib/II Study of Neoadjuvant Chemoradiation Therapy (CRT) Alone or in Combination with Pembrolizumab in Patients with Resectable or Borderline Resectable Pancreatic Cancer. J. Clin. Oncol. 2021, 39, 4128. [Google Scholar] [CrossRef]
- Borazanci, E.H.; Jameson, G.S.; Borad, M.J.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Von Hoff, D.D. A Phase II Pilot Trial of Nivolumab (N) + Albumin Bound Paclitaxel (AP) + Paricalcitol (P) + Cisplatin (C) + Gemcitabine (G) (NAPPCG) in Patients with Previously Untreated Metastatic Pancreatic Ductal Adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36, 358. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of Ipilimumab in Combination with Allogeneic Pancreatic Tumor Cells Transfected with a GM-CSF Gene in Previously Treated Pancreatic Cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Heumann, T.R.; Judkins, C.; Lim, S.J.; Wang, H.; Parkinson, R.; Gai, J.; Celiker, B.; Durham, J.N.; Laheru, D.A.; De Jesus-Acosta, A.; et al. Neoadjuvant and Adjuvant Antitumor Vaccination Alone or Combination with PD1 Blockade and CD137 Agonism in Patients with Resectable Pancreatic Adenocarcinoma. J. Clin. Oncol. 2022, 40, 558. [Google Scholar] [CrossRef]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of Algenpantucel-L Immunotherapy to Standard Adjuvant Therapy for Pancreatic Cancer: A Phase 2 Study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef]
- Hewitt, D.B.; Nissen, N.; Hatoum, H.; Musher, B.; Seng, J.; Coveler, A.L.; Al-Rajabi, R.; Yeo, C.J.; Leiby, B.; Banks, J.; et al. A Phase 3 Randomized Clinical Trial of Chemotherapy with or without Algenpantucel-L (HyperAcute-Pancreas) Immunotherapy in Subjects with Borderline Resectable or Locally Advanced Unresectable Pancreatic Cancer. Ann. Surg. 2022, 275, 45–53. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Vähä-Koskela, M.J.; Heikkilä, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef]
- Rahal, A.; Musher, B.L. Oncolytic viral therapy for pancreatic cancer. J. Surg. Oncol. 2017, 116, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.D.; Monaco, M.L.; Essani, K. The Present Status of Immuno-Oncolytic Viruses in the Treatment of Pancreatic Cancer. Viruses 2020, 12, 1318. [Google Scholar] [CrossRef]
- Ahn, D.H.; Bekaii-Saab, T.S. The Continued Promise and Many Disappointments of Oncolytic Virotherapy in Gastrointestinal Malignancies. Biomedicines 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2019, 26, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Buggy, J.J.; Kortlever, R.; Adimoolam, S.; Monclús, H.A.; Allende, M.T.S.; Swigart, L.B.; Evan, G.I. Modeling Pharmacological Inhibition of Mast Cell Degranulation as a Therapy for Insulinoma. Neoplasia 2011, 13, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.; Javle, M.; Davis, R.E.; Vats, P.; Kumar-Sinha, C.; Xiao, L.; Mettu, N.B.; Parra, E.R.; Benson, A.B.; Lopez, C.D.; et al. Randomized Phase II Study of the Bruton Tyrosine Kinase Inhibitor Acalabrutinib, Alone or with Pembrolizumab in Patients with Advanced Pancreatic Cancer. J. Immunother. Cancer 2020, 8, e000587. [Google Scholar] [CrossRef] [Green Version]
- Di Federico, A.; Tateo, V.; Parisi, C.; Formica, F.; Carloni, R.; Frega, G.; Rizzo, A.; Ricci, D.; Di Marco, M.; Palloni, A.; et al. Hacking Pancreatic Cancer: Present and Future of Personalized Medicine. Pharmaceuticals 2021, 14, 677. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| NCT (Acronym) | Phase | Number of Patients | Setting | Treatment Arms | Primary Endpoint | Status |
|---|---|---|---|---|---|---|
| NCT03989310 | I/II | 50 | Locally advanced/Metastatic | (1) Manganese chloride + Nab-paclitaxel + Gemcitabine + Anti-PD-1 antibody (2) Nab-paclitaxel + Gemcitabine + Anti-PD-1 antibody | Safety DCR | Recruiting |
| NCT04548752 | II | 88 | Maintenance, BRCA mutated | (1) Olaparib + Pembrolizumab 29 Olaparib | PFS | Recruiting |
| NCT04156087 | II | 20 | Locally Advanced | MIS-MWA + Durvalumab + Tremelimumab | PFS | Recruiting |
| NCT05116917 | II | 30 | Metastatic | Nivolumab + Ipilimumab + Influenza vaccine + SBRT | ORR | Recruiting |
| NCT03161379 | II | 30 | Borderline resectable, neoadjuvant | Cyclophosphamide + Nivolumab + GVAX + SBRT | CD8 count (cells/mm3) in the tumor microenvironment | Active, not recruiting |
| NCT04324307 | I/II | 60 | Locally Advanced/ Metastatic | (1) 2nd line PD-L1/CTLA4 inhibitor (2) 1st line PD-L1/CTLA4 inhibitor + gemcitabine/nab-paclitaxel (3) 1st line PD-L1/CTLA4 inhibitor + FOLFIRINOX | ORR | Recruiting |
| NCT03193190 | Ib/II | 290 | Metastatic | Severals, combinations of Nab-Paclitaxel, Gemcitabine, Oxaliplatin, Fluorouracil, Atezolizumab, Cobimetinib, PEGPH20, BL-8040, Selicrelumab, Bevacizumab, RO6874281, AB928, Tiragolumab and Tocilizumab. | ORR Safety | Active, not recruiting |
| NCT04361162 | II | 30 | Metastatic | Nivolumab + Ipilimumab + Radiation | ORR | Recruiting |
| NCT04543071 | II | 10 | Metastatic | Motixafortide, Cemiplimab, Gemcitabine, Nab-Paclitaxel | ORR | Recruiting |
| NCT03336216 | II | 179 | Advanced, pretreated | (1) Gemcitabine/Nab-Paclitaxel or 5-FU/Leucovorin/Irinotecan Liposome (2) Cabiralizumab + Nivolumab (3) Gemcitabine + Nab-Paclitaxel + Cabiralizumab + Nivolumab (4) Cabiralizumab + Nivolumab + FOLFOX | PFS | Active, not recruiting |
| NCT03977272 | III | 110 | Metastatic | (1) modified-FOLFIRINOX/FOLFIRINOX (2) modified-FOLFIRINOX/FOLFIRINOX + anti PD-1 antibody 200 mg | OS | Recruiting |
| NCT03983057 | III | 830 | Locally advanced/borderline resectable | (1) modified-FOLFIRINOX (2) modified-FOLFIRINOX + anti PD-1 antibody 3 mg/kg | PFS | Recruiting |
| NCT04377048 | II | 38 | Metastatic | Tegafur-Gimeracil-Oteracil + Nivolumab + Gemcitabine | ORR | Not yet recruiting |
| NCT04493060 | II | 20 | Metastatic, germline or somatic BRCA1/2 and PALB2 related cancer | Dostarlimab + Niraparib | DCR | Recruiting |
| NCT02648282 | II | 58 | Locally advanced | Cyclophosphamide + Pembrolizumab + GVAX + SBRT | DMFS | Active, not recruiting |
| NCT04887805 | II | 28 | Maintenance after 1st or 2nd line chemotherapy | Lenvatinib + Pembrolizumab | PFS | Recruiting |
| NCT04247165 | I/II | 20 | Locally advanced | Nivolumab + Ipilimumab + Gemcitabine + Nab-paclitaxel + SBRT | Safety | Recruiting |
| NCT05093231 | II | 20 | Metastatic | Pembrolizumab + Olaparib | ORR | Recruiting |
| NCT04940286 | II | 30 | Resectable/borderline resectable, neoadjuvant | Durvalumab + Oleclumab + Nab-paclitaxel + Gemcitabine | Major pathological response rate (≤5% viable tumor cells) Safety | Recruiting |
| NCT02305186 | I/II | 68 | Resectable/borderline resectable, neoadjuvant | (1) chemoradioterapy (with Capecitabine) (2) chemoradioterapy (with Capecitabine) + Pembrolizumab | TILs per HPF Safety | Recruiting |
| NCT04827953 | I/II | 24 | Advanced | Gemcitabine + nab-paclitaxel + NLM-001 + Zalifrelimab | ORR | Recruiting |
| NCT03563248 | II | 160 | Resectable/borderline resectable/locally advanced, neoadjuvant | (1) FOLFIRINOX → SBRT → Surgery (2) FOLFIRINOX + Losartan → SBRT + Losartan → Surgery (3) FOLFIRINOX + Losartan → SBRT + Nivolumab + Losartan → Surgery (4) FOLFIRINOX × 8 → SBRT + Nivolumab → Surgery | Proportion of patients with R0 resection | Recruiting |
| NCT04177810 | II | 21 | Metastatic | Plerixafor + Cemiplimab | ORR | Recruiting |
| NCT05014776 | II | 20 | Metastatic, pretreated | Tadalafil + Pembrolizumab + Ipilimumab + CRS-207 | irORR | Recruiting |
| NCT03190265 | II | 63 | Metastatic, pretreated | (1) Cyclophosphamide + Nivolumab + Ipilimumab + GVAX + CRS-207 (2) Nivolumab + Ipilimumab + CRS-207 | ORR | Active, not recruiting |
| NCT02907099 | IIb | 18 | Metastatic | CXCR4 antagonist BL-8040 + Pembrolizumab | ORR | Active, not recruiting |
| NCT04116073 | II | 25 | Unresectable/Metastatic, pretreated | INCMGA00012 (PD-1 antibody) | DCR4 | Recruiting |
| NCT04753879 | II | 38 | Metastatic | Nab-paclitaxel + Gemcitabine + Cisplatin + Irinotecan + Capecitabine → Maintenance with Pembrolizumab + Olaparib | PFS | Recruiting |
| NCT03767582 | I/II | 30 | Locally advanced | (1) SBRT + Nivolumab + CCR2/CCR5 dual antagonist (2) SBRT + Nivolumab + GVAX + CCR2/CCR5 dual antagonist | Safety Immune response | Recruiting |
| NCT03727880 | II | 36 | Neoadjuvant/Adjuvant, resectable at diagnosis | (1) Pembrolizumab + Defactinib (2) Pembrolizumab | pCR | Recruiting |
| NCT04624217 | Ib/II | 54 | Advanced, pretreated | Gemcitabine + Nab-paclitaxel + SHR-1701 | ORR RP2D | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. https://doi.org/10.3390/cancers14102429
Di Federico A, Mosca M, Pagani R, Carloni R, Frega G, De Giglio A, Rizzo A, Ricci D, Tavolari S, Di Marco M, et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers. 2022; 14(10):2429. https://doi.org/10.3390/cancers14102429
Chicago/Turabian StyleDi Federico, Alessandro, Mirta Mosca, Rachele Pagani, Riccardo Carloni, Giorgio Frega, Andrea De Giglio, Alessandro Rizzo, Dalia Ricci, Simona Tavolari, Mariacristina Di Marco, and et al. 2022. "Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes" Cancers 14, no. 10: 2429. https://doi.org/10.3390/cancers14102429
APA StyleDi Federico, A., Mosca, M., Pagani, R., Carloni, R., Frega, G., De Giglio, A., Rizzo, A., Ricci, D., Tavolari, S., Di Marco, M., Palloni, A., & Brandi, G. (2022). Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers, 14(10), 2429. https://doi.org/10.3390/cancers14102429