
Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia?

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mechanisms and the Landscape of TP53 Alteration
3. Current and Insufficient Standards-of-Care
4. Mutant p53 Protein “Refolding” or “Reactivating” Therapies
5. Leveraging the Immune System
5.1. CD47/SIRPα Axis Inhibition
5.2. Immune Checkpoint Inhibition and Other Immunotherapy
6. MDM2–p53 Interaction Destabilization
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N.; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Montalban-Bravo, G.; Benton, C.B.; Wang, S.A.; Ravandi, F.; Kadia, T.; Cortes, J.; Daver, N.; Takahashi, K.; DiNardo, C.; Jabbour, E.; et al. More than 1 tp53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood 2017, 129, 2584–2587. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerova, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Badar, T.; Atallah, E.; Shallis, R.M.; Goldberg, A.D.; Patel, A.; Abaza, Y.; Bewersdorf, J.P.; Saliba, A.N.; Correia, G.S.C.; Murthy, G.; et al. Outcomes of tp53-mutated aml with evolving frontline therapies: Impact of allogeneic stem cell transplantation on survival. Am. J. Hematol. 2022. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Talati, C.; Desai, P.; Famulare, C.; Devlin, S.M.; Farnoud, N.; Sallman, D.A.; Lancet, J.E.; Roboz, G.J.; Sweet, K.L.; et al. Tp53 mutations predict poorer responses to cpx-351 in acute myeloid leukemia. Blood 2018, 132, 1433. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. Tp53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Middeke, J.M.; Herold, S.; Rucker-Braun, E.; Berdel, W.E.; Stelljes, M.; Kaufmann, M.; Schafer-Eckart, K.; Baldus, C.D.; Stuhlmann, R.; Ho, A.D.; et al. Tp53 mutation in patients with high-risk acute myeloid leukaemia treated with allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2016, 172, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of aml in adults: 2017 eln recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewersdorf, J.P.; Shallis, R.M.; Gowda, L.; Wei, W.; Hager, K.; Isufi, I.; Kim, T.K.; Pillai, M.M.; Seropian, S.; Podoltsev, N.A.; et al. Clinical outcomes and characteristics of patients with tp53-mutated acute myeloid leukemia or myelodysplastic syndromes: A single center experience. Leuk. Lymphoma 2020, 61, 2180–2190. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahme, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt plus azacitidine in tp53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase ii study by the groupe francophone des myelodysplasies (gfm). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (apr-246) and azacitidine in tp53-mutant myelodysplastic syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of tp53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell 2018, 71, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Kayser, S.; Dohner, K.; Krauter, J.; Kohne, C.H.; Horst, H.A.; Held, G.; von Lilienfeld-Toal, M.; Wilhelm, S.; Kundgen, A.; Gotze, K.; et al. The impact of therapy-related acute myeloid leukemia (aml) on outcome in 2853 adult patients with newly diagnosed aml. Blood 2011, 117, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. Tp53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Kern, W.; Kohlmann, A.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosom. Cancer 2005, 43, 227–238. [Google Scholar] [CrossRef]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. Tp53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochazka, K.T.; Pregartner, G.; Rucker, F.G.; Heitzer, E.; Pabst, G.; Wolfler, A.; Zebisch, A.; Berghold, A.; Dohner, K.; Sill, H. Clinical implications of subclonal tp53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of tp53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Seifert, H.; Mohr, B.; Thiede, C.; Oelschlagel, U.; Schakel, U.; Illmer, T.; Soucek, S.; Ehninger, G.; Schaich, M.; Study Alliance, L. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009, 23, 656–663. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Petrenko, O. The mdm2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Quintas-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. P53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of tp53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of tp53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Grob, T.; Al Hinai, A.S.; Sanders, M.A.; Kavelaars, F.; Rijken, M.; Gradowska, P.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk Kooy, M.; et al. Molecular characterization of mutant tp53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Siddon, A.J.; Madanat, Y.; Gagan, J.; Arber, D.A.; Dal Cin, P.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. Tp53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related mds/aml. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; Liu, C.W.; Tang, J.L.; et al. Tp53 mutations in de novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Gibson, C.J.; Murdock, H.M.; Stone, R.M.; Cortes, J.E.; Uy, G.L.; Lin, T.L.; Ritchie, E.K.; Prebet, T.; Ryan, R.J.; et al. Genetic characteristics and outcomes by mutation status in a phase 3 study of cpx-351 versus 7+3 in older adults with newly diagnosed, high-risk/secondary acute myeloid leukemia (aml). Blood 2019, 134, 15. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. Cpx-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Madarang, E.; Lykon, J.; Nguyen, N.; Watts, J.M.; Bradley, T.J.; Chandhok, N.S. Real world outcomes of liposomal daunorubicin and cytarabine versus 7+3 in patients with secondary acute myeloid leukemia. Blood 2020, 136, 5–6. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Dolnik, A.; Tang, L.; Seymour, J.F.; Minden, M.D.; Stone, R.M.; Del Castillo, T.B.; Al-Ali, H.K.; Santini, V.; Vyas, P.; et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia 2018, 32, 2546–2557. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Loghavi, S.; Huang, X.; Qiao, W.; Borthakur, G.; Kadia, T.M.; Daver, N.; Ohanian, M.; Dinardo, C.D.; et al. Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukaemia: A randomised phase 2 trial. Lancet Haematol. 2019, 6, e29–e37. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase iii trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [Green Version]
- Boddu, P.; Kantarjian, H.; Ravandi, F.; Garcia-Manero, G.; Borthakur, G.; Andreeff, M.; Jabbour, E.J.; Benton, C.B.; DiNardo, C.D.; Konopleva, M.; et al. Outcomes with lower intensity therapy in tp53-mutated acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 2238–2241. [Google Scholar] [CrossRef]
- Cashen, A.F.; Schiller, G.J.; O’Donnell, M.R.; DiPersio, J.F. Multicenter, phase ii study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Garzon, R.; Klisovic, R.B.; Schwind, S.; Walker, A.; Geyer, S.; Liu, S.; Havelange, V.; Becker, H.; Schaaf, L.; et al. Clinical response and mir-29b predictive significance in older aml patients treated with a 10-day schedule of decitabine. Proc. Natl. Acad. Sci. USA 2010, 107, 7473–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. Tp53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Zhang, J.; Pillai, R.; Shouse, G.; Sanchez, J.F.; Mei, M.; Nakamura, R.; Stein, A.S.; Forman, S.J.; Marcucci, G.; et al. Venetoclax and hypomethylating agents in tp53-mutated acute myeloid leukaemia. Br. J. Haematol. 2019, 187, e45–e48. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: A single-centre, phase 2 trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- Chiche, E.; Rahme, R.; Bertoli, S.; Dumas, P.Y.; Micol, J.B.; Hicheri, Y.; Pasquier, F.; Peterlin, P.; Chevallier, P.; Thomas, X.; et al. Real-life experience with cpx-351 and impact on the outcome of high-risk aml patients: A multicentric french cohort. Blood Adv. 2021, 5, 176–184. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Podoltsev, N.A.; Wang, X.; Zhang, C.; Bewersdorf, J.P.; Shallis, R.M.; Huntington, S.F.; Neparidze, N.; Giri, S.; Gore, S.D.; et al. Patterns of care and clinical outcomes with cytarabine-anthracycline induction chemotherapy for aml patients in the united states. Blood Adv. 2020, 4, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Ravandi, F.; O’Brien, S.; Cortes, J.; Faderl, S.; Garcia-Manero, G.; Jabbour, E.; Wierda, W.; Kadia, T.; Pierce, S.; et al. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood 2010, 116, 4422–4429. [Google Scholar] [CrossRef]
- Kantarjian, H.; O’Brien, S.; Cortes, J.; Giles, F.; Faderl, S.; Jabbour, E.; Garcia-Manero, G.; Wierda, W.; Pierce, S.; Shan, J.; et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: Predictive prognostic models for outcome. Cancer 2006, 106, 1090–1098. [Google Scholar] [CrossRef]
- Klepin, H.D.; Geiger, A.M.; Tooze, J.A.; Kritchevsky, S.B.; Williamson, J.D.; Pardee, T.S.; Ellis, L.R.; Powell, B.L. Geriatric assessment predicts survival for older adults receiving induction chemotherapy for acute myelogenous leukemia. Blood 2013, 121, 4287–4294. [Google Scholar] [CrossRef]
- Lowenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Milligan, D.; Goldstone, A.; Prentice, A.; McMullin, M.F.; Dennis, M.; Sellwood, E.; Pallis, M.; Russell, N.; Hills, R.K.; et al. The impact of dose escalation and resistance modulation in older patients with acute myeloid leukaemia and high risk myelodysplastic syndrome: The results of the lrf aml14 trial. Br. J. Haematol. 2009, 145, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.H.; Burnett, A.K.; Wheatley, K.; Smith, A.G.; Hutchinson, R.M.; Clark, R.E. Attempts to improve treatment outcomes in acute myeloid leukemia (aml) in older patients: The results of the united kingdom medical research council aml11 trial. Blood 2001, 98, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchner, T.; Berdel, W.E.; Haferlach, C.; Haferlach, T.; Schnittger, S.; Muller-Tidow, C.; Braess, J.; Spiekermann, K.; Kienast, J.; Staib, P.; et al. Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: A study by the german acute myeloid leukemia cooperative group. J. Clin. Oncol. 2009, 27, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Boddu, P.C.; Bewersdorf, J.P.; Zeidan, A.M. The golden age for patients in their golden years: The progressive upheaval of age and the treatment of newly-diagnosed acute myeloid leukemia. Blood Rev. 2020, 40, 100639. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Juliusson, G. Older patients with acute myeloid leukemia benefit from intensive chemotherapy: An update from the swedish acute leukemia registry. Clin. Lymphoma Myeloma Leuk. 2011, 11, S54–S59. [Google Scholar] [CrossRef]
- Juliusson, G.; Antunovic, P.; Derolf, A.; Lehmann, S.; Mollgard, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Hoglund, M. Age and acute myeloid leukemia: Real world data on decision to treat and outcomes from the swedish acute leukemia registry. Blood 2009, 113, 4179–4187. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.B.; Othus, M.; Borthakur, G.; Ravandi, F.; Cortes, J.E.; Pierce, S.A.; Appelbaum, F.R.; Kantarjian, H.A.; Estey, E.H. Prediction of early death after induction therapy for newly diagnosed acute myeloid leukemia with pretreatment risk scores: A novel paradigm for treatment assignment. J. Clin. Oncol. 2011, 29, 4417–4423. [Google Scholar] [CrossRef]
- Appelbaum, F.R.; Gundacker, H.; Head, D.R.; Slovak, M.L.; Willman, C.L.; Godwin, J.E.; Anderson, J.E.; Petersdorf, S.H. Age and acute myeloid leukemia. Blood 2006, 107, 3481–3485. [Google Scholar] [CrossRef]
- Oran, B.; Weisdorf, D.J. Survival for older patients with acute myeloid leukemia: A population-based study. Haematologica 2012, 97, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Wang, R.; Wang, X.; Shallis, R.M.; Podoltsev, N.A.; Bewersdorf, J.P.; Huntington, S.F.; Neparidze, N.; Giri, S.; Gore, S.D.; et al. Clinical outcomes of older patients with aml receiving hypomethylating agents: A large population-based study in the united states. Blood Adv. 2020, 4, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Fenaux, P.; Gobbi, M.; Mayer, J.; Roboz, G.J.; Krauter, J.; Robak, T.; Kantarjian, H.M.; Novak, J.; Jedrzejczaket, W.W. Comparative results of azacitidine and decitabine from a large prospective phase 3 study in treatment naive acute myeloid leukemia (tn-aml) not eligible for intensive therapy. In Proceedings of the European Hematology Association 2020 Meeting, Hamburg, Germany, 12 June 2020; Abstract s142. pp. 11–14. [Google Scholar]
- Short, N.J.; Kantarjian, H.M.; Loghavi, S.; Huang, X.; Qiao, W.; Borthakur, G.; Kadia, T.M.; Daver, N.G.; Ohanian, M.N.; DiNardo, C.D.; et al. Five-day versus ten-day schedules of decitabine in older patients with newly diagnosed acute myeloid leukemia: Results of a randomized phase ii study. Blood 2018, 132, 84. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Pratz, K.W.; Wei, A.H.; Pullarkat, V.A.; Jonas, B.A.; Recher, C.; Babu, S.; Schuh, A.C.; Dail, M.; Sun, Y.; et al. Outcomes in patients with poor-risk cytogenetics with or without tp53 mutations treated with venetoclax combined with hypomethylating agents. In Proceedings of the American Society of Hematology 2021 Meeting, Atlanta, GA, USA, 11 December 2021; Abstract 224. p. 224. [Google Scholar]
- Grenet, J.; Jain, A.G.; Burkart, M.; Waksal, J.; Famulare, C.; Numan, Y.; Stahl, M.; Mckinnell, Z.; Ball, B.; Ma, X.; et al. Comparing outcomes between liposomal daunorubicin/cytarabine (cpx-351) and hma+venetoclax as frontline therapy in acute myeloid leukemia. In Proceedings of the American Society of Hematology 2021 Meeting, Atlanta, GA, USA, 10–14 December 2021; Abstract 32. p. 32. [Google Scholar]
- Ali, D.; Jonsson-Videsater, K.; Deneberg, S.; Bengtzen, S.; Nahi, H.; Paul, C.; Lehmann, S. Apr-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 2011, 86, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of prima-1(met) (apr-246) and 5-azacitidine in tp53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [Green Version]
- Aprea Therapeutics Announces Results of Primary Endpoint from Phase 3 Trial of Eprenetapopt in Tp53 Mutant myelodysPlastic Syndromes (Mds). Aprea Therapeutics. Available online: https://ir.Aprea.Com/news-releases/news-release-details/aprea-therapeutics-announces-results-primary-endpoint-phase-3 (accessed on 28 December 2020).
- Sallman, D.A.; Komrokji, R.S.; Dezern, A.E.; Sebert, M.; Garcia-Manero, G.; Rahmé, R.; Steensma, D.P.; Rahmé, R.; Lehmann, J.; Roboz, G.J.; et al. Long term follow-up and combined phase 2 results of eprenetapopt (apr-246) and azacitidine (aza) in patients with tp53 mutant myelodysplastic syndromes (mds) and oligoblastic acute myeloid leukemia (aml). In Proceedings of the American Society of Hematology 2021 Meeting, Atlanta, GA, USA, 11 December 2021; Abstract 246. p. 246. [Google Scholar]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Gallacher, P.; Wennborg, A.; Kaylor Hickman, D.; et al. Phase ii trial of eprenetapopt (apr-246) in combination with azacitidine (aza) as maintenance therapy for tp53 mutated aml or mds following allogeneic stem cell transplantation (sct). Blood 2021, 138, 409. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Gallacher, P.; Wennborg, A.; Kaylor Hickman, D.; et al. Phase i and expansion study of eprenetapopt (apr-246) in combination with venetoclax (ven) and azacitidine (aza) in tp53-mutant acute myeloid leukemia (aml). Blood 2021, 138, 3409. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xc(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, K.M.; Corrales Benitez, M.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. Slc7a11 is a superior determinant of apr-246 (eprenetapopt) response than tp53 mutation status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. Apr-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022, 107, 403–416. [Google Scholar] [CrossRef]
- Sweeney, C.; Vyas, P. The graft-versus-leukemia effect in aml. Front. Oncol. 2019, 9, 1217. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Wang, J.; Willingham, S.B.; Martin, R.; Wernig, G.; Weissman, I.L. Anti-cd47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012, 26, 2538–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharides, A.P.; Jin, L.; Cheng, P.Y.; Prasolava, T.K.; Malko, A.V.; Ho, J.M.; Poeppl, A.G.; van Rooijen, N.; Minden, M.D.; Danska, J.S.; et al. Disruption of sirpalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 2012, 209, 1883–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The cd47-sirpalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The cd47-signal regulatory protein alpha (sirpa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the cd47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. Cd47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. Cd47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, C.; Li, M.; Xia, B.; Wang, Y.; Zhang, L.; Zhang, Y.; Wang, J.; Sun, F.; Lu, S.; et al. Novel fully human anti-cd47 antibodies stimulate phagocytosis and promote elimination of aml cells. J. Cell. Physiol. 2021, 236, 4470–4481. [Google Scholar] [CrossRef]
- Chen, A.; Harrabi, O.; Fong, A.P.; Ruffner, K.L.; Forgie, A.J.; Sim, J.; Randolph, S.S.; Wan, H.; Pons, J.; Kuo, T.C. Alx148 enhances the depth and durability of response to multiple aml therapies. Blood 2020, 136, 15–16. [Google Scholar] [CrossRef]
- Wang, C.; Sallman, D.A. Targeting the cluster of differentiation 47/signal-regulatory protein alpha axis in myeloid malignancies. Curr. Opin. Hematol. 2022, 29, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The first-in-class anti-cd47 antibody magrolimab (5f9) in combination with azacitidine is effective in mds and aml patients: Ongoing phase 1b results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; Dinardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase i/ii study of azacitidine (aza) with venetoclax (ven) and magrolimab (magro) in patients (pts) with newly diagnosed older/unfit or high-risk acute myeloid leukemia (aml) and relapsed/refractory (r/r) aml. In Proceedings of the American Society of Hematology 2021 Meeting, Atlanta, GA, USA, 12 December 2021; Abstract 371. p. 371. [Google Scholar]
- Chen, X.; Othus, M.; Wood, B.L.; Walter, R.B.; Becker, P.S.; Percival, M.E.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Comparison of myeloid blast counts and variant allele frequencies of gene mutations in myelodysplastic syndrome with excess blasts and secondary acute myeloid leukemia. Leuk. Lymphoma 2021, 62, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Groschel, S.; Schlenk, R.F.; Engelmann, J.; Rockova, V.; Teleanu, V.; Kuhn, M.W.; Eiwen, K.; Erpelinck, C.; Havermans, M.; Lubbert, M.; et al. Deregulated expression of evi1 defines a poor prognostic subset of mll-rearranged acute myeloid leukemias: A study of the german-austrian acute myeloid leukemia study group and the dutch-belgian-swiss hovon/sakk cooperative group. J. Clin. Oncol. 2013, 31, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and safety of sabatolimab (mbg453) in combination with hypomethylating agents (hmas) in patients (pts) with very high/high-risk myelodysplastic syndrome (vhr/hr-mds) and acute myeloid leukemia (aml): Final analysis from a phase ib study. Blood 2021, 138, 244. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. Tp53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in aml. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef]
- Yee, K.; Papayannidis, C.; Vey, N.; Dickinson, M.J.; Kelly, K.R.; Assouline, S.; Kasner, M.; Seiter, K.; Drummond, M.W.; Yoon, S.S.; et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study small star, filled. Leuk. Res. 2021, 100, 106489. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the mdm2 inhibitor amg 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class nedd8-activating enzyme inhibitor, combined with azacitidine in patients with aml. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Peluso, M.O.; Adam, A.; Armet, C.M.; Zhang, L.; O’Connor, R.W.; Lee, B.H.; Lake, A.C.; Normant, E.; Chappel, S.C.; Hill, J.A.; et al. The fully human anti-cd47 antibody srf231 exerts dual-mechanism antitumor activity via engagement of the activating receptor cd32a. J. Immunother. Cancer 2020, 8, e000413. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Erba, H.P.; Sanikommu, S.R.; Altman, J.K.; Sayar, H.; Scott, B.L.; Fond, A.P. Evorpacept (alx148), a cd47-blocking myeloid checkpoint inhibitor, in combination with azacitidine: A phase 1/2 study in patients with myelodysplastic syndrome (aspen-02). In Proceedings of the American Society of Hematology Meeting, Atlanta, GA, USA, 12 December 2021; Abstract 2601. p. 2601. [Google Scholar]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. Tti-621 (sirpalphafc): A cd47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, K.; Sharma, P.; Ali, T.; Khan, I.; Smith, W.C.; MacLeod, A.; Black, C. Measuring the population burden of chronic kidney disease: A systematic literature review of the estimated prevalence of impaired kidney function. Nephrol. Dial. Transplant. 2012, 27, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase iii study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.M.; Advani, A.S.; et al. Phase i study of the cd47 blocker tti-621 in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 2021, 27, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Trillium Therapeutics, Inc. Trillium Therapeutics Provides Data Update, Announces Phase 1b/2 Program Priorities Across Hematologic Malignancies and Solid Tumors, and Reports Governance Changes. Available online: https://s22.Q4cdn.Com/183592819/files/doc_news/2021/04/2021-04-28-rd-day-press-release-final (accessed on 10 June 2021).
- Qi, J.; Li, J.; Jiang, B.; Jiang, B.; Liu, H.; Cao, X.; Zhang, M.; Meng, Y.; MA, X.; Jia, Y.; et al. A phase i/iia study of lemzoparlimab, a monoclonal antibody targeting cd47, in patients with relapsed and/or refractory acute myeloid leukemia (aml) and myelodysplastic syndrome (mds): Initial phase i results. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Shatz, M.; Menendez, D.; Resnick, M.A. The human tlr innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef] [Green Version]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A tale of two stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. P53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. Pd-l1 (b7-h1) and pd-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Nittner, D.; Lambertz, I.; Clermont, F.; Mestdagh, P.; Kohler, C.; Nielsen, S.J.; Jochemsen, A.; Speleman, F.; Vandesompele, J.; Dyer, M.A.; et al. Synthetic lethality between rb, p53 and dicer or mir-17-92 in retinal progenitors suppresses retinoblastoma formation. Nat. Cell Biol. 2012, 14, 958–965. [Google Scholar] [CrossRef]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. P53 represses c-myc through induction of the tumor suppressor mir-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microrna processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. Pdl1 regulation by p53 via mir-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. Tp53 mutations in myelodysplastic syndromes and secondary aml confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Sakuishi, K.; Jayaraman, P.; Behar, S.M.; Anderson, A.C.; Kuchroo, V.K. Emerging tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011, 32, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting tim-3 and pd-1 pathways to reverse t cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A tim-3/gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.Y.; Rollig, C.; Cavenagh, J.; Deeren, D.; Girshova, L.; Krauter, J.; Martinelli, G.; Montesinos, P. A randomized double-blind phase 3 trial of cytarabine with mdm2 inhibitor idasanutlin or placebo in relapsed/refractory acute myeloid leukemia (r/r aml): Primary analysis results of the mirros study. In Proceedings of the 25th Congress of the European Hematology Association 2020, Frankfurt, Germany, 12 June 2020. Abstract ep532. [Google Scholar]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with aml. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.; Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert, E.R.; et al. Pevonedistat and azacitidine upregulate noxa (pmaip1) to increase sensitivity to venetoclax in preclinical models of acute myeloid leukemia. Haematologica 2021, 107, 825–835. [Google Scholar] [CrossRef]
- Short, N.J.; Montalban-Bravo, G.; Alvarado, Y.; Konopleva, M.; Jabbour, E.J.; Garcia-Manero, G.; Yilmaz, M.; Jain, N.; Borthakur, G.; DiNardo, C.D.; et al. Azacitidine, venetoclax and pevonedistat as frontline therapy for patients with secondary acute myeloid leukemia who are unfit for intensive chemotherapy: Results from a phase i/ii study. Blood 2021, 138, 2349. [Google Scholar] [CrossRef]


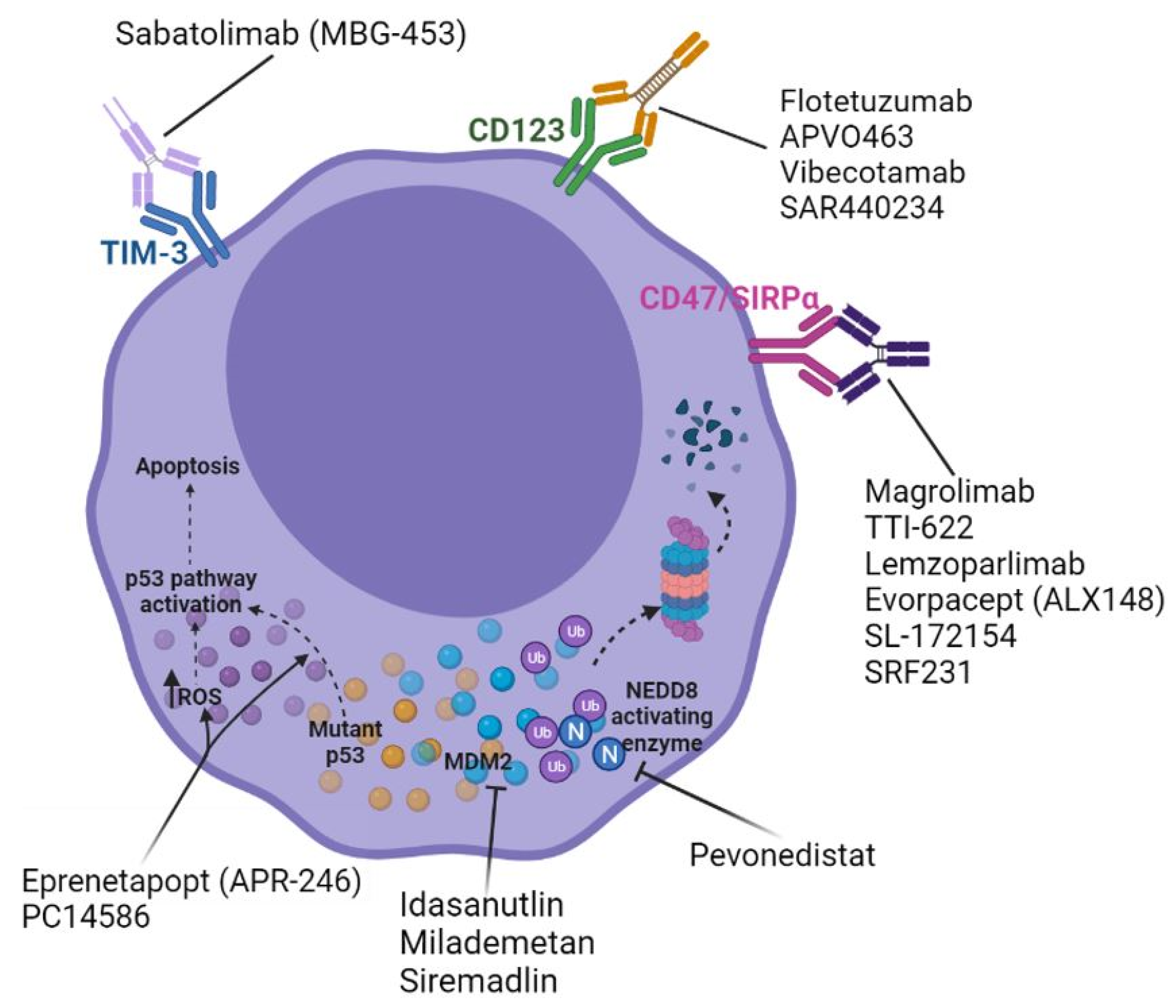{kind=link}
| Regimen | Response Rates | Outcomes | Reference(s) |
|---|---|---|---|
| Intensive induction therapy | |||
| Cytarabine + anthracycline (“7 + 3”) | CR: 28–48% CR/CRi: 33–66% | Median EFS: 1.6–5.7 months Median OS: 5.1–6.5 months | [21,23,24,32,33,34] |
| Liposomal cytarabine + daunorubicin (CPX-351) | CR: 29% CR/CRi: 11–41% | Median EFS: 1.0–8.1 months Median OS: 4.5–8.5 months | [33,34,35] |
| Less-intensive induction therapy | |||
| Azacitidine monotherapy | CR: 40% CR/CRi: 0–40% | Median OS: 7.2 months | [10,36,37] |
| Decitabine monotherapy(5-day schedule) | CR/CRi: 29% | Median OS: 2.1–5.5 months | [38,39,40,41] |
| Decitabine monotherapy(10-day schedule) | CR: 31% CR/CRi: 38–47% | Median EFS: 5.7 months Median OS: 4.9–7.3 months | [38,40,42,43] |
| Azacitidine + venetoclax | CR/CRi: 47–67% | Median EFS: 5.6 months Median OS: 7.2 months | [7,36,44] |
| Decitabine (5-day schedule) + venetoclax | CR/CRi: 47–50% | Median EFS: 5.6 months Median OS: 7.2 months | [7,44] |
| Decitabine (10-day schedule) + venetoclax | CR/CRi: 50–69% | Median EFS: 3.4–5.7 months Median OS: 5.2–6.9 months | [25,44,45] |
| Mechanism of Action | Agent | Interim Clinical Data | References |
|---|---|---|---|
| Mutant p53 “reactivation” | Eprenetapropt (APR-246) | Eprenetapropt + AZA:
| [15,72,89] |
| CD47/SIRPα inhibition | Magrolimab (Hu5F9-G4) | Magrolimab + AZA:
| [90,87] |
| TIM-3 inhibition | Sabatolimab (MBG-453) | Sabatolimab + AZA:
| [91] |
| CD123 x CD3 bispecific antibody therapy | Flotetuzumab | Flotetuzumab monotherapy (R/R population):
| [92] |
| MDM2 inhibition | Idasanutlin | Idasanutlin + cytarabine:
| [93] |
| AMG-232 | AMG-232 +/- trametinib:
| [94] | |
| NEDD8 activating enzyme inhibition | Pevonedistat | Pevonedistat + azacitidine:
| [11,95] |
| Mechanism of Action | Agent | Regimen | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Mutant p53 “reactivation” | Eprenetapropt (APR-246) | Eprenetapropt + azacitidine + venetoclax | NCT04214860 |
| CD47/SIRPα inhibition | SRF213 | Monotherapy | NCT03512340 |
| Evorpacept (ALX148) | Evorpacept + azacitidine + venetoclax | NCT04755244 | |
| TTI-622 | TTI-622 + azacitidine + venetoclax | NCT03530683 | |
| Lemzoparlimab | Lemzoparlimab + azacitidine + venetoclax | NCT04912063 | |
| AK117 | AK117 + azacitidine | NCT04980885 | |
| DSP107 | DSP107 + azacitidine + venetoclax | NCT04937166 | |
| SL-172154 | SL-172154 + azacitidine + venetoclax | NCT05275439 | |
| IBI188 | IBI188 + azacitidine | NCT04485052 | |
| TIM-3 inhibition | Sabatolimab | Sabatolimab + azacitidine + venetoclax | NCT04150029 |
| MDM2 inhibition | Idasanutlin | Idasanutlin + “7 + 3” | NCT03850535 |
| Idasanutlin | Idasanutlin + venetoclax (R/R) | NCT02670044 | |
| Milademetan | Milademetan + low-dose cytarabine +/− venetoclax (R/R) | NCT03634228 | |
| Siremadlin (HDM201) | Siremadlin + azacitidine + venetoclax | NCT05155709 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shallis, R.M.; Bewersdorf, J.P.; Stahl, M.F.; Halene, S.; Zeidan, A.M. Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia? Cancers 2022, 14, 2434. https://doi.org/10.3390/cancers14102434
Shallis RM, Bewersdorf JP, Stahl MF, Halene S, Zeidan AM. Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia? Cancers. 2022; 14(10):2434. https://doi.org/10.3390/cancers14102434
Chicago/Turabian StyleShallis, Rory M., Jan P. Bewersdorf, Maximilian F. Stahl, Stephanie Halene, and Amer M. Zeidan. 2022. "Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia?" Cancers 14, no. 10: 2434. https://doi.org/10.3390/cancers14102434
APA StyleShallis, R. M., Bewersdorf, J. P., Stahl, M. F., Halene, S., & Zeidan, A. M. (2022). Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia? Cancers, 14(10), 2434. https://doi.org/10.3390/cancers14102434