
Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. KIT Mutations in Systemic Mastocytosis
2.1. KIT D816V Mutation
2.2. Other KIT Mutations
3. Clonal Haematopoiesis in Systemic Mastocytosis
4. Mutations in Genes Other Than KIT
4.1. Mutations Affecting Transcription Factors and Signalling Pathways
4.2. Mutations in Genes Involved in Epigenetic Regulatory Mechanisms
4.3. Mutations in Genes Involved in Alternative mRNA Splicing
5. Prognostic Impact of Acquired Gene Mutations in Systemic Mastocytosis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, R.; Bonifacio, M.; Lucchini, G.; Sperr, W.R.; Scaffidi, L.; van Anrooij, B.; Oude Elberink, H.N.; Rossignol, J.; Hermine, O.; Gorska, A.; et al. Refined diagnostic criteria for bone marrow mastocytosis: A proposal of the European competence network on mastocytosis. Leukemia 2022, 36, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Escribano, L.; Alvarez-Twose, I.; Garcia-Montero, A.; Sanchez-Munoz, L.; Jara-Acevedo, M.; Orfao, A. Indolent systemic mastocytosis without skin involvement vs. isolated bone marrow mastocytosis. Haematologica 2011, 96, e26; author reply e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Twose, I.; Jara-Acevedo, M.; Morgado, J.M.; García-Montero, A.; Sánchez-Muñoz, L.; Teodósio, C.; Matito, A.; Mayado, A.; Caldas, C.; Mollejo, M.; et al. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J. Allergy Clin. Immunol. 2016, 137, 168–178.e161. [Google Scholar] [CrossRef]
- Escribano, L.; Alvarez-Twose, I.; Sanchez-Munoz, L.; Garcia-Montero, A.; Nunez, R.; Almeida, J.; Jara-Acevedo, M.; Teodosio, C.; Garcia-Cosio, M.; Bellas, C.; et al. Prognosis in adult indolent systemic mastocytosis: A long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J. Allergy Clin. Immunol. 2009, 124, 514–521. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Advances in the Classification and Treatment of Mastocytosis: Current Status and Outlook toward the Future. Cancer Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Sperr, W.R.; Kundi, M.; Alvarez-Twose, I.; van Anrooij, B.; Oude Elberink, J.N.G.; Gorska, A.; Niedoszytko, M.; Gleixner, K.V.; Hadzijusufovic, E.; Zanotti, R.; et al. International prognostic scoring system for mastocytosis (IPSM): A retrospective cohort study. Lancet Haematol. 2019, 6, e638–e649. [Google Scholar] [CrossRef]
- Longley, B.J.; Tyrrell, L.; Lu, S.Z.; Ma, Y.S.; Langley, K.; Ding, T.G.; Duffy, T.; Jacobs, P.; Tang, L.H.; Modlin, I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: Establishment of clonality in a human mast cell neoplasm. Nat. Genet. 1996, 12, 312–314. [Google Scholar] [CrossRef]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gonzalez, J.I.; Alvarez-Twose, I.; Jara-Acevedo, M.; Henriques, A.; Vinas, E.; Prieto, C.; Sanchez-Munoz, L.; Caldas, C.; Mayado, A.; Matito, A.; et al. Frequency and prognostic impact of KIT and other genetic variants in indolent systemic mastocytosis. Blood 2019, 134, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gonzalez, J.I.; Jara-Acevedo, M.; Alvarez-Twose, I.; Merker, J.D.; Teodosio, C.; Hou, Y.; Henriques, A.; Roskin, K.M.; Sanchez-Munoz, L.; Tsai, A.G.; et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018, 2, 2814–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Orfao, A.; Garcia-Montero, A.C.; Sanchez, L.; Escribano, L. Recent advances in the understanding of mastocytosis: The role of KIT mutations. Br. J. Haematol. 2007, 138, 12–30. [Google Scholar] [CrossRef]
- Li, C.L.; Johnson, G.R. Stem cell factor enhances the survival but not the self-renewal of murine hematopoietic long-term repopulating cells. Blood 1994, 84, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Brown, K.; Qiu, F.H.; Besmer, P. c-kit protein, a transmembrane kinase: Identification in tissues and characterization. Mol. Cell. Biol. 1988, 8, 4896–4903. [Google Scholar] [CrossRef] [Green Version]
- Orfao, A.; Matarraz, S.; Perez-Andres, M.; Almeida, J.; Teodosio, C.; Berkowska, M.A.; van Dongen, J.J.M.; EuroFlow. Immunophenotypic dissection of normal hematopoiesis. J. Immunol. Methods 2019, 475, 112684. [Google Scholar] [CrossRef]
- Teodosio, C.; Mayado, A.; Sanchez-Munoz, L.; Morgado, J.M.; Jara-Acevedo, M.; Alvarez-Twose, I.; Garcia-Montero, A.C.; Matito, A.; Caldas, C.; Escribano, L.; et al. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J. Leukoc. Biol. 2015, 97, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Okayama, Y.; Kawakami, T. Development, migration, and survival of mast cells. Immunol. Res. 2006, 34, 97–115. [Google Scholar] [CrossRef]
- Kitamura, Y.; Hirotab, S. Kit as a human oncogenic tyrosine kinase. Cell. Mol. Life Sci. CMLS 2004, 61, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, H.; Tsujimura, T.; Matsumura, I.; Oritani, K.; Ikeda, H.; Ishikawa, J.; Okabe, M.; Suzuki, M.; Yamamura, K.; Matsuzawa, Y.; et al. Neoplastic transformation of normal hematopoietic cells by constitutively activating mutations of c-kit receptor tyrosine kinase. Blood 1996, 88, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Gonzalez, J.I.; Garcia-Montero, A.C.; Orfao, A.; Alvarez-Twose, I. Pathogenic and diagnostic relevance of KIT in primary mast cell activation disorders. Ann. Allergy Asthma Immunol. 2021, 127, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Metcalfe, D.D. Systemic Mastocytosis. Annu. Rev. Med. 2004, 55, 419–432. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Pullarkat, S.T.; Pullarkat, V.; Kroft, S.H.; Wilson, C.S.; Ahsanuddin, A.N.; Mann, K.P.; Thein, M.; Grody, W.W.; Brynes, R.K. Systemic mastocytosis associated with t(8;21)(q22;q22) acute myeloid leukemia. J. Hematop. 2009, 2, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Sotlar, K.; Horny, H.P.; Simonitsch, I.; Krokowski, M.; Aichberger, K.J.; Mayerhofer, M.; Printz, D.; Fritsch, G.; Valent, P. CD25 indicates the neoplastic phenotype of mast cells: A novel immunohistochemical marker for the diagnosis of systemic mastocytosis (SM) in routinely processed bone marrow biopsy specimens. Am. J. Surg. Pathol. 2004, 28, 1319–1325. [Google Scholar] [CrossRef]
- Pignon, J.M.; Giraudier, S.; Duquesnoy, P.; Jouault, H.; Imbert, M.; Vainchenker, W.; Vernant, J.P.; Tulliez, M. A new c-kit mutation in a case of aggressive mast cell disease. Br. J. Haematol. 1997, 96, 374–376. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Elala, Y.; Wassie, E.; Finke, C.; Reichard, K.K.; Chen, D.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Next-generation sequencing in systemic mastocytosis: Derivation of a mutation-augmented clinical prognostic model for survival. Am. J. Hematol. 2016, 91, 888–893. [Google Scholar] [CrossRef]
- Baek, J.O.; Kang, H.K.; Na, S.Y.; Lee, J.R.; Roh, J.Y.; Lee, J.H.; Kim, H.J.; Park, S. N822K c-kit mutation in CD30-positive cutaneous pleomorphic mastocytosis after germ cell tumour of the ovary. Br. J. Dermatol. 2012, 166, 1370–1373. [Google Scholar] [CrossRef]
- Arredondo, A.R.; Gotlib, J.; Shier, L.; Medeiros, B.; Wong, K.; Cherry, A.; Corless, C.; Arber, D.A.; Valent, P.; George, T.I. Myelomastocytic leukemia versus mast cell leukemia versus systemic mastocytosis associated with acute myeloid leukemia: A diagnostic challenge. Am. J. Hematol. 2010, 85, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Alvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lubke, J.; Perkins, C.; Munoz-Gonzalez, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Cabral do, O.H.N.; Naumann, N.; Jawhar, M.; Weiss, C.; Metzgeroth, G.; Schmid, A.; Lubke, J.; Reiter, L.; Fabarius, A.; et al. Importance of Adequate Diagnostic Workup for Correct Diagnosis of Advanced Systemic Mastocytosis. J. Allergy Clin. Immunol. Pract. 2020, 8, 3121–3127.e3121. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.A.; Pullarkat, S.T.; Calverley, D.C.; Brynes, R.K. Mast cell disease associated with acute myeloid leukemia: Detection of a new c-kit mutation Asp816His. Am. J. Hematol. 2000, 65, 307–309. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Bueso-Ramos, C.; Lai, R.; Kroft, S.; Wilson, C.S.; Pullarkat, S.T.; Bu, X.; Thein, M.; Lee, M.; Brynes, R.K. Systemic mastocytosis with associated clonal hematological non-mast-cell lineage disease: Analysis of clinicopathologic features and activating c-kit mutations. Am. J. Hematol. 2003, 73, 12–17. [Google Scholar] [CrossRef]
- Sotlar, K.; Colak, S.; Bache, A.; Berezowska, S.; Krokowski, M.; Bultmann, B.; Valent, P.; Horny, H.P. Variable presence of KITD816V in clonal haematological non-mast cell lineage diseases associated with systemic mastocytosis (SM-AHNMD). J. Pathol. 2010, 220, 586–595. [Google Scholar] [CrossRef]
- Longley, B.J., Jr.; Metcalfe, D.D.; Tharp, M.; Wang, X.; Tyrrell, L.; Lu, S.Z.; Heitjan, D.; Ma, Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 1609–1614. [Google Scholar] [CrossRef] [Green Version]
- Horny, H.P.; Sotlar, K.; Sperr, W.R.; Valent, P. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: A histopathological challenge. J. Clin. Pathol. 2004, 57, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Nagai, S.; Ichikawa, M.; Takahashi, T.; Sato, H.; Yokota, H.; Oshima, K.; Izutsu, K.; Hangaishi, A.; Kanda, Y.; Motokura, T.; et al. The origin of neoplastic mast cells in systemic mastocytosis with AML1/ETO-positive acute myeloid leukemia. Exp. Hematol. 2007, 35, 1747–1752. [Google Scholar] [CrossRef]
- Lasho, T.; Finke, C.; Zblewski, D.; Hanson, C.A.; Ketterling, R.P.; Butterfield, J.H.; Tefferi, A.; Pardanani, A. Concurrent activating KIT mutations in systemic mastocytosis. Br. J. Haematol. 2016, 173, 153–156. [Google Scholar] [CrossRef]
- Yabe, M.; Masukawa, A.; Kato, S.; Yabe, H.; Nakamura, N.; Matsushita, H. Systemic mastocytosis associated with t(8;21) acute myeloid leukemia in a child: Detection of the D816A mutation of KIT. Pediatric Blood Cancer 2012, 59, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, M.; Miura, H.; Inagaki, H.; Shinkai, Y.; Kato, A.; Kato, T.; Hamada-Tsutsumi, S.; Tanaka, M.; Kudo, K.; Yoshikawa, T.; et al. An aggressive systemic mastocytosis preceded by ovarian dysgerminoma. BMC Cancer 2020, 20, 1162. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Chakrabarti, S.; Akin, C.; Robyn, J.; Bahceci, E.; Greene, A.; Childs, R.; Dunbar, C.E.; Metcalfe, D.D.; Barrett, A.J. A pilot study of nonmyeloablative allogeneic hematopoietic stem cell transplant for advanced systemic mastocytosis. Bone Marrow Transpl. 2006, 37, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Joensuu, H.; Demetri, G.D.; Corless, C.L.; Apperley, J.; Fletcher, J.A.; Soulieres, D.; Dirnhofer, S.; Harlow, A.; Town, A.; et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin. Cancer Res. 2008, 14, 2717–2725. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, J.K.; Shao, L.; Bixby, D.L.; Ross, C.W. Shared clonal cytogenetic abnormalities in aberrant mast cells and leukemic myeloid blasts detected by single nucleotide polymorphism microarray-based whole-genome scanning. Genes Chromosomes Cancer 2016, 55, 389–396. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Meggendorfer, M.; Naumann, N.; Horny, H.P.; Sotlar, K.; Haferlach, T.; Schmitt, K.; Fabarius, A.; Valent, P.; et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica 2017, 102, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Lanternier, F.; Cohen-Akenine, A.; Palmerini, F.; Feger, F.; Yang, Y.; Zermati, Y.; Barète, S.; Sans, B.; Baude, C.; Ghez, D.; et al. Phenotypic and Genotypic Characteristics of Mastocytosis According to the Age of Onset. PLoS ONE 2008, 3, e1906. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Berger, J.; Cerny-Reiterer, S.; Peter, B.; Eisenwort, G.; Hoermann, G.; Mullauer, L.; Mannhalter, C.; Steurer, M.; Bettelheim, P.; et al. Chronic mast cell leukemia (MCL) with KIT S476I: A rare entity defined by leukemic expansion of mature mast cells and absence of organ damage. Ann. Hematol. 2015, 94, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Georgin-Lavialle, S.; Lhermitte, L.; Suarez, F.; Yang, Y.; Letard, S.; Hanssens, K.; Feger, F.; Renand, A.; Brouze, C.; Canioni, D.; et al. Mast cell leukemia: Identification of a new c-Kit mutation, dup(501-502), and response to masitinib, a c-Kit tyrosine kinase inhibitor. Eur. J. Haematol. 2012, 89, 47–52. [Google Scholar] [CrossRef]
- Rouet, A.; Aouba, A.; Damaj, G.; Soucie, E.; Hanssens, K.; Chandesris, M.O.; Livideanu, C.B.; Dutertre, M.; Durieu, I.; Grandpeix-Guyodo, C.; et al. Mastocytosis among elderly patients: A multicenter retrospective French study on 53 patients. Medicine 2016, 95, e3901. [Google Scholar] [CrossRef]
- Soucie, E.; Hanssens, K.; Mercher, T.; Georgin-Lavialle, S.; Damaj, G.; Livideanu, C.; Chandesris, M.O.; Acin, Y.; Létard, S.; de Sepulveda, P.; et al. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 2012, 120, 4846–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mital, A.; Piskorz, A.; Lewandowski, K.; Wasag, B.; Limon, J.; Hellmann, A. A case of mast cell leukaemia with exon 9 KIT mutation and good response to imatinib. Eur. J. Haematol. 2011, 86, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Smith, M.L.; Schultheis, B.; Fitzgibbon, J.; Lister, T.A.; Melo, J.V.; Cross, N.C.; Cavenagh, J.D. A novel K509I mutation of KIT identified in familial mastocytosis-in vitro and in vivo responsiveness to imatinib therapy. Leuk. Res. 2006, 30, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Fumo, G.; Yavuz, A.S.; Lipsky, P.E.; Neckers, L.; Metcalfe, D.D. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood 2004, 103, 3222–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, V.; Waghorn, K.; Langabeer, S.E.; Jeffers, M.; Cross, N.C.P.; Hayden, P.J. Molecular response to imatinib in KIT F522C-mutated systemic mastocytosis. Leuk. Res. 2019, 77, 28–29. [Google Scholar] [CrossRef]
- Nakagomi, N.; Hirota, S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab. Investig. 2007, 87, 365–371. [Google Scholar] [CrossRef]
- Büttner, C.; Henz, B.M.; Welker, P.; Sepp, N.T.; Grabbe, J. Identification of Activating c-kit Mutations in Adult-, but not in Childhood-Onset Indolent Mastocytosis: A Possible Explanation for Divergent Clinical Behavior. J. Investig. Dermatol. 1998, 111, 1227–1231. [Google Scholar] [CrossRef]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y.; et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef]
- Spector, M.S.; Iossifov, I.; Kritharis, A.; He, C.; Kolitz, J.E.; Lowe, S.W.; Allen, S.L. Mast-cell leukemia exome sequencing reveals a mutation in the IgE mast-cell receptor beta chain and KIT V654A. Leukemia 2012, 26, 1422–1425. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach-Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef]
- Wang, H.J.; Lin, Z.M.; Zhang, J.; Yin, J.H.; Yang, Y. A new germline mutation in KIT associated with diffuse cutaneous mastocytosis in a Chinese family. Clin. Exp. Dermatol. 2014, 39, 146–149. [Google Scholar] [CrossRef] [PubMed]
- de Melo Campos, P.; Machado-Neto, J.A.; Scopim-Ribeiro, R.; Visconte, V.; Tabarroki, A.; Duarte, A.S.; Barra, F.F.; Vassalo, J.; Rogers, H.J.; Lorand-Metze, I.; et al. Familial systemic mastocytosis with germline KIT K509I mutation is sensitive to treatment with imatinib, dasatinib and PKC412. Leuk. Res. 2014, 38, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Twose, I.; Matito, A.; Morgado, J.M.; Sanchez-Munoz, L.; Jara-Acevedo, M.; Garcia-Montero, A.; Mayado, A.; Caldas, C.; Teodosio, C.; Munoz-Gonzalez, J.I.; et al. Imatinib in systemic mastocytosis: A phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget 2017, 8, 68950–68963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, M.J.; Ferrao, P.T.; Hughes, T.P.; Ashman, L.K. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol. Cancer Ther. 2002, 1, 1115–1124. [Google Scholar] [PubMed]
- Akin, C.; Brockow, K.; D’Ambrosio, C.; Kirshenbaum, A.S.; Ma, Y.; Longley, B.J.; Metcalfe, D.D. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp. Hematol. 2003, 31, 686–692. [Google Scholar] [CrossRef]
- Ma, Y.; Zeng, S.; Metcalfe, D.D.; Akin, C.; Dimitrijevic, S.; Butterfield, J.H.; McMahon, G.; Longley, B.J. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood 2002, 99, 1741–1744. [Google Scholar] [CrossRef]
- Valent, P.; Blatt, K.; Eisenwort, G.; Herrmann, H.; Cerny-Reiterer, S.; Thalhammer, R.; Müllauer, L.; Hoermann, G.; Sadovnik, I.; Schwarzinger, I.; et al. FLAG-induced remission in a patient with acute mast cell leukemia (MCL) exhibiting t(7;10)(q22;q26) and KIT D816H. Leuk. Res. Rep. 2014, 3, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lubke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.P.; Sotlar, K.; et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer 2018, 57, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Horny, H.P.; Escribano, L.; Longley, B.J.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nunez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- Yavuz, A.S.; Lipsky, P.E.; Yavuz, S.; Metcalfe, D.D.; Akin, C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood 2002, 100, 661–665. [Google Scholar] [CrossRef]
- Akin, C. Clonality and molecular pathogenesis of mastocytosis. Acta Haematol. 2005, 114, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, C.N.; Yavuz, A.S.; Lipsky, P.E.; Metcalfe, D.D.; Akin, C. Analysis of the lineage relationship between mast cells and basophils using the c-kit D816V mutation as a biologic signature. J. Allergy Clin. Immunol. 2005, 115, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Kirshenbaum, A.S.; Semere, T.; Worobec, A.S.; Scott, L.M.; Metcalfe, D.D. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp. Hematol. 2000, 28, 140–147. [Google Scholar] [CrossRef]
- Mayado, A.; Teodosio, C.; Dasilva-Freire, N.; Jara-Acevedo, M.; Garcia-Montero, A.C.; Alvarez-Twose, I.; Sanchez-Munoz, L.; Matito, A.; Caldas, C.; Munoz-Gonzalez, J.I.; et al. Characterization of CD34(+) hematopoietic cells in systemic mastocytosis: Potential role in disease dissemination. Allergy 2018, 73, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Bock, C.; George, T.I.; Galli, S.J.; Gotlib, J.; Haferlach, T.; Hoermann, G.; Hermine, O.; et al. Proposed Terminology and Classification of Pre-Malignant Neoplastic Conditions: A Consensus Proposal. EBioMedicine 2017, 26, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.L.; Sehgal, D.; Raffeld, M.; Obiakor, H.; Akin, C.; Mage, R.G.; Metcalfe, D.D. Demonstration that mast cells, T cells, and B cells bearing the activating kit mutation D816V occur in clusters within the marrow of patients with mastocytosis. J. Mol. Diagn. JMD 2004, 6, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Alvarez-Twose, I.; Teodosio, C.; Sanchez-Munoz, L.; Muniz, C.; Munoz-Gonzalez, J.I.; Mayado, A.; Matito, A.; Caldas, C.; et al. KIT D816V-mutated bone marrow mesenchymal stem cells in indolent systemic mastocytosis are associated with disease progression. Blood 2016, 127, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Jara-Acevedo, M.; Teodosio, C.; Sanchez-Munoz, L.; Alvarez-Twose, I.; Mayado, A.; Caldas, C.; Matito, A.; Morgado, J.M.; Munoz-Gonzalez, J.I.; Escribano, L.; et al. Detection of the KIT D816V mutation in peripheral blood of systemic mastocytosis: Diagnostic implications. Mod. Pathol. 2015, 28, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Teodosio, C.; Garcia-Montero, A.C.; Jara-Acevedo, M.; Sanchez-Munoz, L.; Pedreira, C.E.; Alvarez-Twose, I.; Matarraz, S.; Morgado, J.M.; Barcena, P.; Matito, A.; et al. Gene expression profile of highly purified bone marrow mast cells in systemic mastocytosis. J. Allergy Clin. Immunol. 2013, 131, 1213–1224. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS ONE 2012, 7, e43090. [Google Scholar] [CrossRef]
- Damaj, G.; Joris, M.; Chandesris, O.; Hanssens, K.; Soucie, E.; Canioni, D.; Kolb, B.; Durieu, I.; Gyan, E.; Livideanu, C.; et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suffering systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS ONE 2014, 9, 85362. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Szankasi, P.; Sederberg, M.; Schumacher, J.; Frizzell, K.A.; Gee, E.P.; Patel, J.L.; South, S.T.; Xu, X.; Kelley, T.W. Concurrent detection of targeted copy number variants and mutations using a myeloid malignancy next generation sequencing panel allows comprehensive genetic analysis using a single testing strategy. Br. J. Haematol. 2016, 173, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lubke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.M.; Maric, I.; Simakova, O.; Bai, Y.; Chan, E.C.; Olivares, N.; Carter, M.; Maric, D.; Robyn, J.; Metcalfe, D.D. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica 2011, 96, 459–463. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Muller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Visconte, V.; Makishima, H.; Maciejewski, J.P.; Tiu, R.V. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia 2012, 26, 2447–2454. [Google Scholar] [CrossRef] [Green Version]
- Hanssens, K.; Brenet, F.; Agopian, J.; Georgin-Lavialle, S.; Damaj, G.; Cabaret, L.; Chandesris, M.O.; de Sepulveda, P.; Hermine, O.; Dubreuil, P.; et al. SRSF2-p95 hotspot mutation is highly associated with advanced forms of mastocytosis and mutations in epigenetic regulator genes. Haematologica 2014, 99, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Levine, R.L.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.Y.; Pardanani, A.; et al. Frequent TET2 mutations in systemic mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009, 23, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Lasho, T.L.; Finke, C.; Zblewski, D.L.; Abdelrahman, R.A.; Wassie, E.A.; Gangat, N.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. ASXL1 and CBL mutations are independently predictive of inferior survival in advanced systemic mastocytosis. Br. J. Haematol. 2016, 175, 534–536. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3699. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017, 31, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Haenisch, B.; Frohlich, H.; Herms, S.; Molderings, G.J. Evidence for contribution of epigenetic mechanisms in the pathogenesis of systemic mast cell activation disease. Immunogenetics 2014, 66, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Hausmann, D.; Clemens, J.; Naumann, N.; Henzler, T.; Horny, H.P.; Sotlar, K.; Schoenberg, S.O.; Cross, N.C.; et al. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia 2016, 30, 2342–2350. [Google Scholar] [CrossRef]
- Munoz-Gonzalez, J.I.; Alvarez-Twose, I.; Jara-Acevedo, M.; Zanotti, R.; Perkins, C.; Jawhar, M.; Sperr, W.R.; Shoumariyeh, K.; Schwaab, J.; Greiner, G.; et al. Proposed global prognostic score for systemic mastocytosis: A retrospective prognostic modelling study. Lancet Haematol 2021, 8, e194–e204. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Leardini, D.; Messelodi, D.; Muratore, E.; Baccelli, F.; Bertuccio, S.N.; Anselmi, L.; Pession, A.; Masetti, R. Role of CBL Mutations in Cancer and Non-Malignant Phenotype. Cancers 2022, 14, 839. [Google Scholar] [CrossRef]
- Chung, Y.R.; Schatoff, E.; Abdel-Wahab, O. Epigenetic alterations in hematopoietic malignancies. Int. J. Hematol. 2012, 96, 413–427. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, D.N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019, 573, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nature reviews. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, D.S. Transcription-Factor Mutations and Disease. N. Engl. J. Med. 1996, 334, 28–33. [Google Scholar] [CrossRef]
- Vainchenker, W.; Delhommeau, F.; Constantinescu, S.N.; Bernard, O.A. New mutations and pathogenesis of myeloproliferative neoplasms. Blood 2011, 118, 1723–1735. [Google Scholar] [CrossRef]
- Kales, S.C.; Ryan, P.E.; Nau, M.M.; Lipkowitz, S. Cbl and human myeloid neoplasms: The Cbl oncogene comes of age. Cancer Res. 2010, 70, 4789–4794. [Google Scholar] [CrossRef] [Green Version]
- Sargin, B.; Choudhary, C.; Crosetto, N.; Schmidt, M.H.; Grundler, R.; Rensinghoff, M.; Thiessen, C.; Tickenbrock, L.; Schwable, J.; Brandts, C.; et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 2007, 110, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, M.A.; Briesewitz, R.; Yu, J.; Wang, L.; Wei, M.; Arnoczky, K.J.; Marburger, T.B.; Wen, J.; Perrotti, D.; Bloomfield, C.D.; et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 2007, 110, 1022–1024. [Google Scholar] [CrossRef] [Green Version]
- Reindl, C.; Quentmeier, H.; Petropoulos, K.; Greif, P.A.; Benthaus, T.; Argiropoulos, B.; Mellert, G.; Vempati, S.; Duyster, J.; Buske, C.; et al. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clin. Cancer Res. 2009, 15, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef] [Green Version]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, H.; Cazzolli, H.; Szpurka, H.; Dunbar, A.; Tiu, R.; Huh, J.; Muramatsu, H.; O’Keefe, C.; Hsi, E.; Paquette, R.L.; et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J. Clin. Oncol. 2009, 27, 6109–6116. [Google Scholar] [CrossRef] [Green Version]
- Bader, M.S.; Meyer, S.C. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals 2022, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N Am. 2021, 35, 217–236. [Google Scholar] [CrossRef]
- Naumann, N.; Lubke, J.; Shomali, W.; Reiter, L.; Horny, H.P.; Jawhar, M.; Dangelo, V.; Fabarius, A.; Metzgeroth, G.; Kreil, S.; et al. Clinical and histopathological features of myeloid neoplasms with concurrent Janus kinase 2 (JAK2) V617F and KIT proto-oncogene, receptor tyrosine kinase (KIT) D816V mutations. Br. J. Haematol. 2021, 194, 344–354. [Google Scholar] [CrossRef]
- Chiosea, S.I.; Sherer, C.K.; Jelic, T.; Dacic, S. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod. Pathol. 2011, 24, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Krasinskas, A.M.; Moser, A.J.; Saka, B.; Adsay, N.V.; Chiosea, S.I. KRAS mutant allele-specific imbalance is associated with worse prognosis in pancreatic cancer and progression to undifferentiated carcinoma of the pancreas. Mod. Pathol. 2013, 26, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.Y.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jeng, K.J.; Yang, S.H.; Wang, H.S.; Lan, Y.T.; Lin, C.C.; Liang, W.Y.; et al. Impact of KRAS mutation on outcome of patients with metastatic colorectal cancer. Hepato Gastroenterol. 2014, 61, 1946–1953. [Google Scholar]
- Pardanani, A.; Shah, S.; Mannelli, F.; Elala, Y.C.; Guglielmelli, P.; Lasho, T.L.; Patnaik, M.M.; Gangat, N.; Ketterling, R.P.; Reichard, K.K.; et al. Mayo alliance prognostic system for mastocytosis: Clinical and hybrid clinical-molecular models. Blood Adv. 2018, 2, 2964–2972. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, M.; Goyama, S.; Asai, T.; Kawazu, M.; Nakagawa, M.; Takeshita, M.; Chiba, S.; Ogawa, S.; Kurokawa, M. AML1/Runx1 Negatively Regulates Quiescent Hematopoietic Stem Cells in Adult Hematopoiesis. J. Immunol. 2008, 180, 4402–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Harada, Y.; Imagawa, J.; Kimura, A.; Harada, H. AML1/RUNX1 point mutation possibly promotes leukemic transformation in myeloproliferative neoplasms. Blood 2009, 114, 5201–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Rock, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [Green Version]
- Jawhar, M.; Schwaab, J.; Naumann, N.; Horny, H.-P.; Sotlar, K.; Haferlach, T.; Metzgeroth, G.; Fabarius, A.; Valent, P.; Hofmann, W.-K.; et al. Response and progression on midostaurin in advanced systemic mastocytosis: KIT D816V and other molecular markers. Blood 2017, 130, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Lasho, T.; Barraco, D.; Patnaik, M.; Elala, Y.; Tefferi, A. Next generation sequencing of myeloid neoplasms with eosinophilia harboring the FIP1L1-PDGFRA mutation. Am. J. Hematol. 2016, 91, 10–11. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Reszka, E.; Jablonska, E.; Wieczorek, E.; Valent, P.; Arock, M.; Nilsson, G.; Nedoszytko, B.; Niedoszytko, M. Epigenetic Changes in Neoplastic Mast Cells and Potential Impact in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 2964. [Google Scholar] [CrossRef]
- Gorska, A.; Jablonska, E.; Reszka, E.; Niedoszytko, M.; Lange, M.; Gruchala-Niedoszytko, M.; Jarczak, J.; Strapagiel, D.; Gorska-Ponikowska, M.; Bastian, P.; et al. DNA methylation profile in patients with indolent systemic mastocytosis. Clin. Transl. Allergy 2021, 11, e12074. [Google Scholar] [CrossRef]
- Katoh, M. Functional and cancer genomics of ASXL family members. Br. J. Cancer 2013, 109, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsi-Boyer, V.; Trouplin, V.; Adelaide, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Carbuccia, N.; Murati, A.; Trouplin, V.; Brecqueville, M.; Adelaide, J.; Rey, J.; Vainchenker, W.; Bernard, O.A.; Chaffanet, M.; Vey, N.; et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009, 23, 2183–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.A.; Jankowska, A.; Makishima, H.; Visconte, V.; Jerez, A.; Sugimoto, Y.; Muramatsu, H.; Traina, F.; Afable, M.; Guinta, K.; et al. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica 2013, 98, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010, 24, 1128–1138. [Google Scholar] [CrossRef]
- Jia, Y.; Li, P.; Fang, L.; Zhu, H.; Xu, L.; Cheng, H.; Zhang, J.; Li, F.; Feng, Y.; Li, Y.; et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016, 2, 16007. [Google Scholar] [CrossRef]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Tie, R.; Zhang, T.; Fu, H.; Wang, L.; Wang, Y.; He, Y.; Wang, B.; Zhu, N.; Fu, S.; Lai, X.; et al. Association between DNMT3A mutations and prognosis of adults with de novo acute myeloid leukemia: A systematic review and meta-analysis. PLoS ONE 2014, 9, e93353. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.; Cross, N.C.P. Aberrations of EZH2 in Cancer. Clinical Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.L.T.M.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet 2010, 42, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Holmfeldt, L.; Mullighan, C.G. The role of TET2 in hematologic neoplasms. Cancer Cell 2011, 20, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Bejar, R. Splicing Factor Mutations in Cancer. Adv. Exp. Med. Biol. 2016, 907, 215–228. [Google Scholar] [CrossRef]
- Fu, X.D. Specific commitment of different pre-mRNAs to splicing by single SR proteins. Nature 1993, 365, 82–85. [Google Scholar] [CrossRef]
- Edmond, V.; Moysan, E.; Khochbin, S.; Matthias, P.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011, 30, 510–523. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Roller, A.; Haferlach, T.; Eder, C.; Dicker, F.; Grossmann, V.; Kohlmann, A.; Alpermann, T.; Yoshida, K.; Ogawa, S.; et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012, 120, 3080–3088. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Loffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kolking, B.; Wichmann, M.; Gorlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, S.A.; Smith, A.E.; Kulasekararaj, A.G.; Kizilors, A.; Mohamedali, A.M.; Lea, N.C.; Mitsopoulos, K.; Ford, K.; Nasser, E.; Seidl, T.; et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica 2013, 98, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [Green Version]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [Green Version]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet 2011, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, S.; Flotho, C.; Moetter, J.; Heuser, M.; Hasle, H.; Gruhn, B.; Klingebiel, T.; Thol, F.; Schlegelberger, B.; Baumann, I.; et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood 2012, 119, e96–e99. [Google Scholar] [CrossRef] [Green Version]
- Broesby-Olsen, S.; Kristensen, T.K.; Møller, M.B.; Bindslev-Jensen, C.; Vestergaard, H. Adult-onset systemic mastocytosis in monozygotic twins with KIT D816V and JAK2 V617F mutations. J. Allergy Clin. Immunol. 2012, 130, 806–808. [Google Scholar] [CrossRef]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Galata, G.; Garcia-Montero, A.C.; Kristensen, T.; Dawoud, A.A.Z.; Munoz-Gonzalez, J.I.; Meggendorfer, M.; Guglielmelli, P.; Hoade, Y.; Alvarez-Twose, I.; Gieger, C.; et al. Genome-wide association study identifies novel susceptibility loci for KIT D816V positive mastocytosis. Am. J. Hum. Genet. 2021, 108, 284–294. [Google Scholar] [CrossRef] [PubMed]
- De Vita, S.; Schneider, R.K.; Garcia, M.; Wood, J.; Gavillet, M.; Ebert, B.L.; Gerbaulet, A.; Roers, A.; Levine, R.L.; Mullally, A.; et al. Loss of Function of TET2 Cooperates with Constitutively Active KIT in Murine and Human Models of Mastocytosis. PLoS ONE 2014, 9, 96209. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, T.; Vestergaard, H.; Moller, M.B. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J. Mol. Diagn. JMD 2011, 13, 180–188. [Google Scholar] [CrossRef]
- Kristensen, T.; Vestergaard, H.; Bindslev-Jensen, C.; Moller, M.B.; Broesby-Olsen, S. Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am. J. Hematol. 2014, 89, 493–498. [Google Scholar] [CrossRef]
- Erben, P.; Schwaab, J.; Metzgeroth, G.; Horny, H.P.; Jawhar, M.; Sotlar, K.; Fabarius, A.; Teichmann, M.; Schneider, S.; Ernst, T.; et al. The KIT D816V expressed allele burden for diagnosis and disease monitoring of systemic mastocytosis. Ann. Hematol. 2014, 93, 81–88. [Google Scholar] [CrossRef]
- Hoermann, G.; Gleixner, K.V.; Dinu, G.E.; Kundi, M.; Greiner, G.; Wimazal, F.; Hadzijusufovic, E.; Mitterbauer, G.; Mannhalter, C.; Valent, P.; et al. The KIT D816V allele burden predicts survival in patients with mastocytosis and correlates with the WHO type of the disease. Allergy 2014, 69, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Hoermann, G.; Sotlar, K.; Jawhar, M.; Kristensen, T.; Bachelot, G.; Nedoszytko, B.; Carter, M.C.; Horny, H.P.; Bonadonna, P.; Sperr, W.R.; et al. Standards of Genetic Testing in the Diagnosis and Prognostication of Systemic Mastocytosis in 2022: Recommendations of the EU-US Cooperative Group. J. Allergy Clin. Immunol. Pract. 2022. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.J.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Al Hafidh, J.; Balmert, E.; Dabbas, B.; Vaupel, C.; El Hader, C.; McGinniss, M.; Beruti, S.; Bejar, R. Somatic Mutations Indicative of Clonal Hematopoiesis Are Present in a Large Fraction of Cytopenic Patients Who Lack Diagnostic Evidence of MDS. Blood 2014, 124, 3272. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Kern, W.; Hoermann, G.; Milosevic Feenstra, J.D.; Sotlar, K.; Pfeilstöcker, M.; Germing, U.; Sperr, W.R.; Reiter, A.; Wolf, D.; et al. Clonal Hematopoiesis with Oncogenic Potential (CHOP): Separation from CHIP and Roads to AML. Int. J. Mol. Sci. 2019, 20, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Pardanani, A.; Elala, Y.C.; Lasho, T.L.; Patnaik, M.M.; Reichard, K.K.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. Cytogenetic abnormalities in systemic mastocytosis: WHO subcategory-specific incidence and prognostic impact among 348 informative cases. Am. J. Hematol. 2018, 93, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youk, J.; Koh, Y.; Kim, J.W.; Kim, D.Y.; Park, H.; Jung, W.J.; Ahn, K.S.; Yun, H.; Park, I.; Sun, C.H.; et al. A scientific treatment approach for acute mast cell leukemia: Using a strategy based on next-generation sequencing data. Blood Res. 2016, 51, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Biancon, G.; Patel, T.; Pan, Z.; Kothari, S.; Halene, S.; Prebet, T.; Xu, M.L. Comprehensive Clinicopathologic and Molecular Analysis of Mast Cell Leukemia With Associated Hematologic Neoplasm: A Report and In-Depth Study of 5 Cases. Front. Oncol. 2021, 11, 730503. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Zblewski, D.; Patnaik, M.; Ketterling, R.P.; Chen, D.; Hanson, C.A.; Tefferi, A.; Pardanani, A. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J. 2015, 5, e275. [Google Scholar] [CrossRef] [Green Version]
- Toledo, M.A.S.; Gatz, M.; Sontag, S.; Gleixner, K.V.; Eisenwort, G.; Feldberg, K.; Hamouda, A.E.I.; Kluge, F.; Guareschi, R.; Rossetti, G.; et al. Nintedanib targets KIT D816V neoplastic cells derived from induced pluripotent stem cells of systemic mastocytosis. Blood 2021, 137, 2070–2084. [Google Scholar] [CrossRef]
- Dorrance, A. “Mast”ering drug discovery with iPSCs. Blood 2021, 137, 1993–1994. [Google Scholar] [CrossRef]
- Shomali, W.; Gotlib, J. Response Criteria in Advanced Systemic Mastocytosis: Evolution in the Era of KIT Inhibitors. Int. J. Mol. Sci. 2021, 22, 2983. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Andersohn, F.; Konzen, C.; Garbe, E. Systematic review: Agranulocytosis induced by nonchemotherapy drugs. Ann. Intern. Med. 2007, 146, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.B.; Metcalfe, D.D.; Miller, J.S.; Earl, H.; Sullivan, T. Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. N. Engl. J. Med. 1987, 316, 1622–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, L.B. Diagnostic value of tryptase in anaphylaxis and mastocytosis. Immunol. Allergy Clin. N. Am. 2006, 26, 451–463. [Google Scholar] [CrossRef] [PubMed]


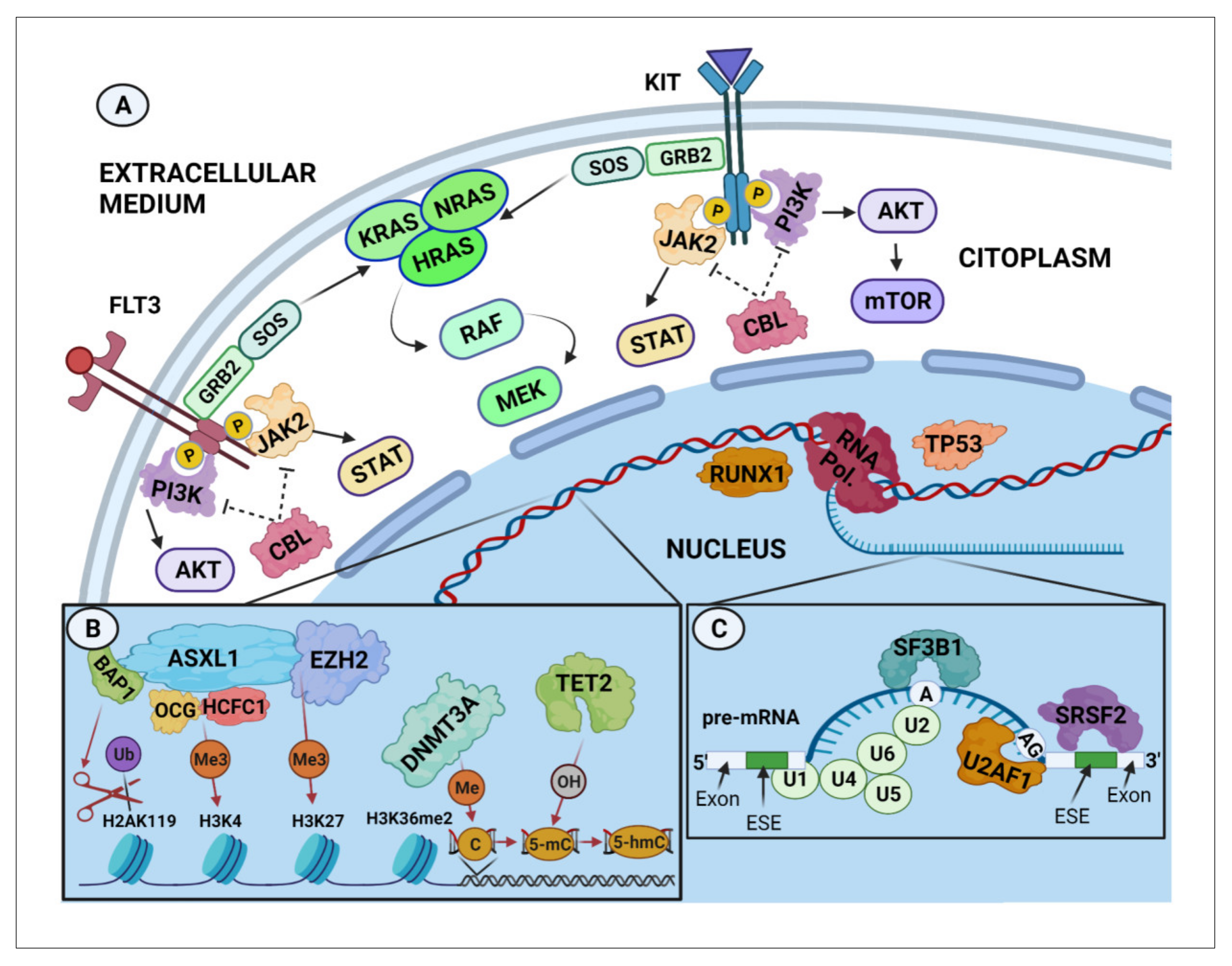{kind=link}
| Domain | Exon | Mutation | Subtype SM |
|---|---|---|---|
| Extracellular: Ligand (SCF) binding domain | 2 | R49C | SM-u [29] |
| 5 | Y269C | SM-AHN [40] | |
| Extracellular: Dimerization domain | 7 | V399I | SM-u [29] |
| 8 | D419del | ISM [47] | |
| 9 | S451C | SM-u [61] | |
| S476I | MCL [48] | ||
| S501_A502dup | ASM [50] MCL [49] | ||
| A502_Y503dup | SM-u [29] MCL [51,52] | ||
| Y503_F504InsAY | ASM [40] | ||
| F504_N505delIns5 | SM-AHN [51] | ||
| K509R | SM-u [29] | ||
| K509I | ISM [62] ASM [53] MCL [62] WDSM [63] | ||
| Transmembrane domain | 10 | F522C | WDSM [32,54,55] MCL [46] |
| Juxta-membrane domain | 11 | V559I | ASM [56] |
| V560G | SM-u [40] ISM [57] MCL [58] | ||
| TK1 domain | 13 | K642E | ASM [29,40] |
| V654A | MCL [59] | ||
| TK2 domain | 17 | A814S | SM-AHN [26] |
| A814T | SM-AHN [27] | ||
| I815-V816Ins | ISM [9] | ||
| D816H | AdvSM [32,33] SM-AHN [26,34,35,36] MCL [46,67,68] | ||
| D816Y | ISM [37] AdvSM [32,33] SM-AHN [26,27,36,38,39] MCL [9,46] | ||
| D816I | SM-AHN [40] | ||
| D816A | SM-AHN [41,45] ASM [42] | ||
| D816G | MCL [43] | ||
| D816T | SM-u [44] | ||
| I817V | WDSM [9] | ||
| D820G | ASM [28] | ||
| N822K | SM-u [30] SM-AHN [31] | ||
| 18 | V852G | SM-u [29] |
| Gene | Exon | Gene Mutations | ||||
|---|---|---|---|---|---|---|
| ASXL1 | 6 | S135C [12] | ||||
| 8 | G219V [12] | |||||
| 12 | Y591* [80] | I641fs [13] | P698Afs* [32] | I919Yfs* [29] | H1008Tfs* [29] | |
| E602* [29] | G642fs [12] | R786Efs* [29] | P920Tfs* [80] | G1026Dfs* [29] | ||
| A611T [29] | G643Wfs* [13] | D820Mfs* [29] | R965_G966del [12] | I1220F [80] | ||
| I617Pfs* [32] | G646Wfs* [29,81] | T844fs [12] | Y974* [32] | G1397S [29] | ||
| H630fs [12]/Gfs* [29] | G646Afs* [29] | S846Vfs* [81] | I980Kfs* [32] | A1521S [12] | ||
| E635Rfs* [13,29,32] | R693* [13] | P849Lfs* [29] | E997* [29] | *1542fs [13] | ||
| CBL | 8 | Q367dup [80] | Y371C [29]/H [29]/S [29] | M374K [29] | L380P [10,29,82] | |
| C384R [12]/Y [29] | M400K [10,32] | C404Y [10,29] | W408C [10,32] | |||
| 9 | G413D [29] | R420Q [10,29] | I423N [29] | |||
| 11 | R550W [12] | |||||
| DNMT3A | 3 | E30A [80] | ||||
| 4 | N90S [12] | |||||
| 8 | R320* [12,29] | |||||
| 10 | A380V [12] | K420* [12] | ||||
| 15 | W581C [12] | L594Cfs* [29] | R598* [29] | D600Afs* [29] | ||
| 16 | S638C [12] | |||||
| 17 | S663L [12] | |||||
| 18 | S714F [12] | R720L [12] | ||||
| 19 | E733G [29] | F755S [12] | R771G [29]/Q [80] | |||
| 23 | N879D [13] | R882C [12,13]/H [12,13,80] | ||||
| EZH2 | 3 | L50Wfs* [13] | ||||
| 5 | I146T [13] | |||||
| 7 | S220F [32] | |||||
| 8 | R288* [32] | |||||
| 14 | Q545* [13] | |||||
| 15 | R583Q [13] | N608K [13] | ||||
| 17 | F672L [29] | |||||
| 18 | R690C [29] | H694R [40] | ||||
| JAK2 | 14 | V617F [12,29,32] | S605Y [12] | |||
| KRAS | 2 | V14I [83] | ||||
| 3 | Q70H [12] | |||||
| 5 | I187N [12] | |||||
| NRAS | 2 | G12S [29] | G12D [29,84] | G13D [84] | ||
| 3 | Q61L [29] | |||||
| RUNX1 | 4 | L56S [29] | P86fs [13] | E88Rfs* [13] | S94I [29] | |
| D96Gfs [85] | R107C [13] | N109T [13]/del [29] | F116L [12] | |||
| 5 | S141L [29] | A142T [13] | N146K [12,13] | R162K [13,29] | ||
| 7 | V238Gfs* [13] | |||||
| SF3B1 | 5 | Y141C [12] | ||||
| 14 | R625C [29] | W658C [29] | T663I [29] | K666N [29]/T [13,32] | ||
| 15 | K700E [13,29,83] | A711D [86] | ||||
| SRSF2 | 1 | V18L [10] | P95 A [13]/H [10,32,85]/L [10]/R [12,13]/T [87] | |||
| TET2 | 3 | L34F [29] | Q321* [83] | P562Tfs* [29] | Q729* [32] | Q933* [51] |
| H192Y [13] | E368* [13] | N595Ifs* [51] | Q731* [51] | Q939* [29] | ||
| V218Wfs* [32] | Q373Rfs* [51] | P612fs [12] | Q734* [51] | K948Nfs* [83] | ||
| Y234* [51] | P409Lfs* [80] | L615Sfs* [29] | Q752_fs* [29] | W954* [29] | ||
| R248Q [29] | G429R [29] | Y620fs [12] | L757Tfs* [29] | Q958Tfs* [29] | ||
| S254Rfs* [51] | L431* [51] | Y634* [29] | L806Rfs* [29] | Q963* [29] | ||
| E259Gfs* [29] | E452Rfs* [29,88] | Q652* [29] | Q810* [29,88] | S972Ffs* [29] | ||
| N275Ifs* [12,13] | D527Gfs* [29] | Q652Sfs* [80] | N837Yfs* [32] | C1016Wfs* [29,88] | ||
| Q278* [29] | Q530* [29] | Q659Rfs* [29] | L840* [51] | Q1020* [80] | ||
| T279fs [12] | E537Sfs* [29] | Q684Nfs* [29] | T849Hfs* [89] | I1024Qfs* [51] | ||
| N281* [29] | R550* [29] | Q705Sfs* [29] | Y867H [29] | P1061Qfs* [51] | ||
| R282G [29] | H558Lfs* [51] | A727S [12] | V872Cfs* [29] | Q1084P [29] | ||
| 4 | D1143Mfs* [32,51] | |||||
| 5 | Q1170* [88] | |||||
| 6 | H1219D [32] | Y1245Lfs [85] | S1246* [51] | Y1255fs [12] | ||
| 8 | Y1337* [80] | A1341E [85] | ||||
| 9 | Y1351* [51] | R1359 C [88]/H [29] | S1369L [29] | H1380Y [29] | D1384V [29] | |
| Q1389* [51] | T1393I [29] | |||||
| 10 | R1465* [29] | R1467G_fs* [29] | K1493fs [12] | |||
| 11 | L1515Ffs* [51] | M1615* [89] | V1718L [29] | L1819* [29] | N1890S [12] | |
| L1531A_fs* [29,88] | Q1652Hfs* [29] | P1723S [29] | I1873T [80] | R1891G [29] | ||
| K1533* [29] | Y1679L_fs* [29] | D1750Efs* [51] | E1879* [29] | F1901Lfs* [51] | ||
| E1555R_fs* [29] | Q1680* [29] | N1765* [29] | H1881L [29]/R [29,51,88] | Y1902C [29] | ||
| Y1598Sfs* [29] | S1688_fs* [29,51,88] | M1800Dfs* [51] | T1884A [29] | H1904R [80] | ||
| S1611Y [12] | M1701I [29] | H1817Pfs* [29] | L1886S [29] | H1912Y [13] | ||
| Gene | SM Diagnostic Subgroup | Mutated Cases/Total Cases (%) | Overall Frequency | WHO Subtype | Mutated Cases/ Total Cases (%) | Overall Frequency | |
|---|---|---|---|---|---|---|---|
| CBL | Non-AdvSM | 1/12 (0) [10] 1/216 (0.5) [12] 0/6 (0) [13] 0/44 (0) [29] 0/1 (0) [50] 1/29 (3.4) [51] 0/26 (0) [68] 0/15 (0) [80] 0/6 (0) [85] | 1% | BMM | 0/65 (0) [12] | 0% | |
| ISM | 1/10 (10) [10] 0/3 (0) [13] 0/1 (0) [50] 0/26 (0) [68] 0/4 (0) [85] | 1/144 (1) [12] 0/44 (0) [29] 1/28 (4) [51] 0/15 (0) [80] | 1% | ||||
| SSM | 0/2 (0) [10] 0/3 (0) [13] 0/2 (0) [85] | 0/7 (0) [12] 0/1 (0) [51] | 0% | ||||
| AdvSM | 7/27 (26) [10] 1/13 (8) [12] 0/14 (0) [13] 16/106 (15) [29] 25/272 (9) [32] 1/25 (4) [50] 0/35 (0) [51] 10/83 (12) [68] 1/10 (10) [80] 3/26 (12) [81] 2/13 (15) [85] 4/19 (21) [90] | 11% | ASM | 0/1 (0) [10] 0/9 (0) [13] 0/2 (0) [50] 0/3 (0) [68] 0/1 (0) [85] | 0/9 (0) [12] 1/25 (4) [29] 0/9 (0) [51] 0/2 (0) [80] 0/6 (0) [90] | 2% | |
| SM-AHN | 6/23 (26) [10] 0/5 (0) [13] 1/21 (5) [50] 10/72 (14) [68] 2/12 (17) [85] | 1/4 (25) [12] 15/80 (19) [29] 0/23 (0) [51] 3/26 (12) [81] 4/13 (31) [90] | 15% | ||||
| MCL | 2/7 (29) [10] 0/2 (0) [50] 0/8 (0) [68] | 0/1 (0) [29] 0/3 (0) [51] | 10% | ||||
| JAK2 | Non-AdvSM | 0/12 (0) [10] 2/97 (2) [12] 0/6 (0) [13] 0/44 (0) [29] 0/1 (0) [50] 0/29 (0) [51] 2/26 (8) [68] 0/6 (0) [85] 0/13 (0) [88] | 2% | BMM | 1/23 (4) [12] | 4% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/1 (0) [50] 2/26 (8) [68] 0/13 (0) [88] | 1/70 (1) [12] 0/44 (0) [29] 0/28 (0) [51] 0/4 (0) [85] | 2% | ||||
| SSM | 0/2 (0) [10] 0/3 (0) [13] 0/2 (0) [85] | 0/4 (0) [12] 0/1 (0) [51] | 0% | ||||
| AdvSM | 2/27 (7) [10] 0/14 (0) [13] 9/106 (9) [29] 25/213 (12) [32] 3/25 (12) [50] 3/35 (9) [51] 12/83 (15) [68] 3/47 (6) [81] 1/13 (8) [85] 2/29 (7) [88] | 10% | ASM | 0/1 (0) [10] 0/9 (0) [13] 0/2 (0) [50] 0/3 (0) [68] 0/5 (0) [88] | 0/7 (0) [12] 0/25 (0) [29] 0/9 (0) [51] 0/1 (0) [85] | 0% | |
| SM-AHN | 2/23 (9) [10] 0/5 (0) [13] 3/21 (14) [50] 12/72 (17) [68] 1/12 (8) [85] | 0/3 (0) [12] 9/80 (11) [29] 3/23 (13) [51] 3/47 (6) [81] 2/23 (9) [88] | 11% | ||||
| MCL | 0/3 (0) [10] 0/1 (0) [50] 0/8 (0) [68] | 0/1 (0) [29] 0/3 (0) [51] 0/1 (0) [88] | 0% | ||||
| KRAS | Non-AdvSM | 0/12 (0) [10] 2/97 (2) [12] 0/6 (0) [13] 0/29 (0) [51] 0/36 (0) [84] | 1% | BMM | 0/23 (0) [12] | 0% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/27 (0) [84] | 2/70 (3) [12] 0/28 (0) [51] | 1% | ||||
| SSM | 0/2 (0) [10] 0/3 (0) [13] 0/9 (0) [84] | 0/4 (0) [12] 0/1 (0) [51] | 0% | ||||
| AdvSM | 4/27 (15) [10] 0/10 (0) [12] 0/14 (0) [13] 0/35 (0) [51] 2/16 (13) [68] | 6% | ASM | 0/1 (0) [10] 0/9 (0) [13] | 0/7 (0) [12] 0/9 (0) [51] | 0% | |
| SM-AHN | 4/23 (17) [10] 0/5 (0) [13] 2/16 (13) [68] | 0/3 (0) [12] 0/23 (0) [51] | 9% | ||||
| MCL | 0/3 (0) [10] | 0/2 (0) [51] | 0% | ||||
| NRAS | Non-AdvSM | 0/12 (0) [10] 0/6 (0) [13] 0/44 (0) [29] 0/1 (0) [50] 0/23 (0) [51] 0/36 (0) [84] 1/298 (0.3) [97] | 0.2% | BMM | |||
| ISM | 0/10 (0) [10] 0/44 (0) [29] 0/22 (0) [51] | 0/3 (0) [13] 0/1 (0) [50] 0/27 (0) [84] | 0% | ||||
| SSM | 0/2 (0) [10] 0/1 (0) [51] | 0/3 (0) [13] 0/9 (0) [84] | 0% | ||||
| AdvSM | 2/27 (7) [10] 0/14 (0) [13] 3/105 (3) [29] 1/25 (4) [50] 1/25 (4) [51] 2/16 (13) [68] 3/173 (2) [97] | 3% | ASM | 0/1 (0) [10] 0/25 (0) [29] 0/7 (0) [51] | 0/9 (0) [13] 0/2 (0) [50] | 0% | |
| SM-AHN | 2/23 (9) [10] 3/80 (4) [29] 1/16 (6) [51] | 0/5 (0) [13] 1/21 (5) [50] 2/16 (13) [68] | 6% | ||||
| MCL | 0/3 (0) [10] 0/2 (0) [50] | 1/1 (100) [29] 0/2 (0) [51] | 13% | ||||
| RUNX1 | Non-AdvSM | 0/12 (0) [10] 1/309 (0.3) [12] 2/10 (20) [13] 0/44 (0) [29] 0/26 (0) [68] 0/6 (0) [85] 1/530 (0.2) [97] | 0.4% | BMM | 1/90 (1) [12] | 1% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/26 (0) [68] | 0/211 (0) [12] 0/44 (0) [29] 0/4 (0) [85] | 0% | ||||
| SSM | 0/2 (0) [10] 2/7 (29) [13] | 0/8 (0) [12] 0/2 (0) [85] | 11% | ||||
| AdvSM | 9/27 (33) [10] 0/13 (0) [12] 7/24 (29) [13] 5/106 (5) [29] 66/329 (20) [32] 15/83 (18) [68] 1/13 (8) [85] 38/210 (18) [97] | 18% | ASM | 1/1 (100) [10] 2/11 (18) [13] 1/3 (33) [68] | 0/9 (0) [12] 0/25 (0) [29] 0/1 (0) [85] | 8% | |
| SM-AHN | 8/23 (35) [10] 5/13 (39) [13] 14/72 (19) [68] | 0/4 (0) [12] 5/80 (6) [29] 1/12 (8) [85] | 16% | ||||
| MCL | 0/3 (0) [10] 0/8 (0) [68] | 0/1 (0) [29] | 0% | ||||
| Gene | SM Diagnostic Subgroup | Mutated Cases/Total Cases (%) | Overall Frequency | WHO Subtype | Mutated Cases/ Total Cases (%) | Overall Frequency | |
|---|---|---|---|---|---|---|---|
| ASXL1 | Non-AdvSM | 0/12 (0) [10] 6/309 (2) [12] 0/10 (0) [13] 0/44 (0) [29] 0/1 (0) [50] 0/26 (0) [68] 1/15 (7) [80] 0/6 (0) [85] 6/530 (1) [97] | 1% | BMM | 1/90 (1) [12] | 1% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/1 (0) [50] 1/15 (7) [80] 6/530 (1) [97] | 4/211 (2) [12] 0/44 (0) [29] 0/26 (0) [68] 0/4 (0) [85] | 1% | ||||
| SSM | 0/2 (0) [10] 0/7 (0) [13] | 1/8 (13) [12] 0/2 (0) [85] | 5% | ||||
| AdvSM | 8/27 (30) [10] 2/13 (15) [12] 2/24 (8) [13] 25/106 (24) [29] 66/229 (29) [32] 12/25 (48) [50] 21/83 (25) [68] 2/10 (20) [80] 6/43 (14) [81] 5/13 (39) [85] 5/19 (26) [90] 35/210 (17) [97] | 24% | ASM | 0/1 (0) [10] 1/11 (9) [13] 0/2 (0) [50] 0/2 (0) [80] 1/6 (17) [90] | 1/9 (11) [12] 4/25 (16) [29] 0/3 (0) [68] 0/1 (0) [85] | 9% | |
| SM-AHN | 8/23 (35) [10] 1/13 (8) [13] 4/21 (19) [50] 6/43 (14) [81] 4/13 (31) [90] | 1/4 (25) [12] 21/80 (26) [29] 21/72 (29) [68] 5/12 (42) [85] | 25% | ||||
| MCL | 0/3 (0) [10] 0/2 (0) [50] | 0/1 (0) [29] 0/8 (0) [68] | 0% | ||||
| DNMT3A | Non-AdvSM | 14/309 (5) [12] 0/10 (0) [13] 2/44 (5) [29] 0/26 (0) [68] 2/15 (13) [80] 20/530 (4) [97] | 4% | BMM | 2/90 (2) [12] | 2% | |
| ISM | 10/211 (0.5) [12] 2/44 (5) [29] 2/15 (13) [80] | 0/3 (0) [13] 0/26 (0) [68] | 5% | ||||
| SSM | 2/8 (25) [12] | 0/7 (0) [13] | 13% | ||||
| AdvSM | 4/13 (31) [12] 3/24 (13) [13] 7/106 (7) [29] 1/83 (1) [68] 1/10 (10) [80] 2/19 (11) [90] 9/210 (4) [97] | 6% | ASM | 3/9 (33) [12] 0/25 (0) [29] 0/2 (0) [80] | 3/11 (27) [13] 0/3 (0) [68] 0/6 (0) [90] | 11% | |
| SM-AHN | 1/4 (25) [12] 7/80 (9) [29] 1/8 (13) [80] | 0/13 (0) [13] 1/72 (1) [68] 2/13 (15) [90] | 6% | ||||
| MCL | 0/1 (0) [29] | 0/8 (0) [68] | 0% | ||||
| EZH2 | Non-AdvSM | 0/12 (0) [10] 0/309 (0) [12] 1/10 (10) [13] 0/44 (0) [29] 0/26 (0) [68] 0/15 (0) [80] 0/6 (0) [85] | 0.2% | BMM | 0/90 (0) [12] | 0% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/26 (0) [68] 0/4 (0) [85] | 0/211 (0) [12] 0/44 (0) [29] 0/15 (0) [80] | 0% | ||||
| SSM | 0/2 (0) [10] 1/7 (14) [13] | 0/8 (0) [12] 0/2 (0) [85] | 5% | ||||
| AdvSM | 2/27 (7) [10] 0/13 (0) [12] 4/24 (17) [13] 3/106 (3) [29] 17/305 (6) [32] 2/83 (2) [68] 0/10 (0) [80] 2/13 (15) [85] | 5% | ASM | 0/1 (0) [10] 2/11 (18) [13] 0/3 (0) [68] 0/1 (0) [85] | 0/9 (0) [12] 1/25 (4) [29] 0/2 (0) [80] | 6% | |
| SM-AHN | 2/23 (9) [10] 2/13 (15) [13] 2/72 (3) [68] 2/12 (17) [85] | 0/4 (0) [12] 2/80 (3) [29] 0/8 (0) [80] | 5% | ||||
| MCL | 0/3 (0) [10] 0/8 (0) [68] | 0/1 (0) [29] | 0% | ||||
| TET2 | Non-AdvSM | 0/12 (0) [10] 7/309 (2) [12] 0/10 (0) [13] 0/1 (0) [50] 2/29 (7) [51] 1/26 (4) [68] 1/15 (7) [80] 0/6 (0) [85] 2/13 (15) [88] | 3% | BMM | 2/90 (2) [12] | 2% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 1/28 (4) [51] 1/15 (7) [80] 2/13 (15) [88] | 5/211 (2) [12] 0/1 (0) [50] 1/26 (4) [68] 0/4 (0) [85] | 3% | ||||
| SSM | 0/2 (0) [10] 0/7 (0) [13] 0/2 (0) [85] | 0/8 (0) [12] 1/1 (100) [51] | 5% | ||||
| AdvSM | 15/27 (56) [10] 2/13 (15) [12] 3/24 (13) [13] 137/329 (42) [32] 12/25 (48) [50] 12/35 (34) [51] 33/83 (40) [68] 5/10 (50) [80] 12/32 (38) [81] 6/13 (46) [85] 10/29 (35) [88] 5/19 (26) [90] | 39% | ASM | 0/1 (0) [10] 0/11 (0) [13] 3/9 (33) [51] 0/2 (0) [80] 2/5 (40) [88] | 1/9 (11) [12] 2/2 (100) [50] 0/3 (0) [68] 0/1 (0) [85] 1/6 (17) [90] | 21% | |
| SM-AHN | 15/23 (65) [10] 3/13 (23) [13] 9/23 (39) [51] 5/8 (63) [80] 6/12 (50) [85] 4/13 (31) [90] | 1/4 (25) [12] 9/21 (43) [50] 33/72 (46) [68] 12/32 (38) [81] 8/23 (35) [88] | 43% | ||||
| MCL | 0/3 (0) [10] 0/3 (0) [51] 0/1 (0) [88] | 1/2 (50) [50] 0/8 (0) [68] | 6% | ||||
| Gene | SM Prognostic Subgroup | Mutated Cases/ Total Cases (%) | Overall Frequency | WHO Subtype | Mutated Cases/ Total Cases (%) | Overall Frequency | |
|---|---|---|---|---|---|---|---|
| SF3B1 | Non-AdvSM | 2/309 (0.6) [12] 0/10 (0) [13] 0/44 (0) [29] 0/26 (0) [68] 0/6 (0) [85] | 0.5% | BMM | 1/90 (1) [12] | 1% | |
| ISM | 1/211 (0.5) [12] 0/44 (0) [29] 10/4 (0) [85] | 0/3 (0) [13] 0/26 (0) [68] | 0.3% | ||||
| SSM | 0/8 (0) [12] 0/2 (0) [85] | 0/7 (0) [13] | 0% | ||||
| AdvSM | 0/13 (0) [12] 3/24 (13) [13] 9/106 (9) [29] 18/305 (6) [32] 2/83 (2) [68] 1/13 (8) [85] | 7% | ASM | 0/9 (0) [12] 1/25 (4) [29] 0/1 (0) [85] | 2/11 (18) [13] 0/3 (0) [68] | 6% | |
| SM-AHN | 0/4 (0) [12] 7/80 (9) [29] 1/12 (8) [85] | 1/13 (8) [13] 2/72 (3) [68] | 6% | ||||
| MCL | 1/1 (100) [29] | 0/8 (0) [68] | 13% | ||||
| SRSF2 | Non-AdvSM | 0/12 (0) [10] 0/309 (0) [12] 0/10 (0) [13] 0/44 (0) [29] 0/1 (0) [50] 0/26 (0) [68] 0/6 (0) [85] 7/530 (1) [97] | 0.7% | BMM | 0/90 (0) [12] | 0% | |
| ISM | 0/10 (0) [10] 0/3 (0) [13] 0/1 (0) [50] 0/4 (0) [85] | 0/211 (0) [12] 0/44 (0) [29] 0/26 (0) [68] | 0% | ||||
| SSM | 0/2 (0) [10] 0/7 (0) [13] | 0/8 (0) [12] 0/2 (0) [85] | 0% | ||||
| AdvSM | 14/27 (52) [10] 2/13 (15) [12] 3/24 (13) [13] 1/106 (1) [29] 120/329 (37) [32] 8/25 (32) [50] 31/83 (37) [68] 4/13 (31) [85] 79/210 (38) [97] | 32% | ASM | 0/1 (0) [10] 0/11 (0) [13] 1/2 (50) [50] 0/1 (0) [85] | 1/9 (11) [12] 0/25 (0) [29] 0/3 (0) [68] | 4% | |
| SM-AHN | 13/23 (57) [10] 3/13 (23) [13] 7/21 (33) [50] 4/12 (33) [85] | 1/4 (25) [12] 1/80 (1) [29] 31/72 (43) [68] | 27% | ||||
| MCL | 1/3 (33) [10] 0/2 (0) [50] | 0/1 (0) [29] 0/8 (0) [68] | 7% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-López, O.; Muñoz-González, J.I.; Orfao, A.; Álvarez-Twose, I.; García-Montero, A.C. Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis. Cancers 2022, 14, 2487. https://doi.org/10.3390/cancers14102487
González-López O, Muñoz-González JI, Orfao A, Álvarez-Twose I, García-Montero AC. Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis. Cancers. 2022; 14(10):2487. https://doi.org/10.3390/cancers14102487
Chicago/Turabian StyleGonzález-López, Oscar, Javier I. Muñoz-González, Alberto Orfao, Iván Álvarez-Twose, and Andrés C. García-Montero. 2022. "Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis" Cancers 14, no. 10: 2487. https://doi.org/10.3390/cancers14102487
APA StyleGonzález-López, O., Muñoz-González, J. I., Orfao, A., Álvarez-Twose, I., & García-Montero, A. C. (2022). Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis. Cancers, 14(10), 2487. https://doi.org/10.3390/cancers14102487