2. Materials and Methods
2.1. Vectors and Vaccine Production
The recombinant thioredoxin (TRX, 13 kDa), vimentin (Vim, 55 kDa), and TRXtr-Vim (61 kDa) proteins were produced and purified as previously described [
9]. The protein-coding sequences of all three proteins were cloned into the multiple cloning site of the pET21a(+) (Novagen, Merck Millipore, Darmstadt, Germany) expression vector (
Figure S1A–C). Reference sequences were retrieved from UniProt: POAA25-1 for TRX and P20152-1 for Vim. The pET21-TRX plasmid was transformed into
E. coli strain Rosetta Gami (DE3) (Novagen, Merck Millipore), while the pET21-Vim and pET21-TRXtr-Vim vectors were transformed into
E. coli strain BL21 (DE3) (Novagen, Merck Millipore) and stored as glycerol stocks at −80 °C.
LB medium containing 100 µg/mL ampicillin (Amp, Sigma Aldrich, Zwijnsdrecht, The Netherlands, Cat. A9518-5G) was inoculated with bacterial stocks and grown overnight at 37 °C at 200 rpm. The next day, cultures were diluted 1:3 with LB medium containing Amp, and protein expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG, Serva, Heidelberg, Germany, Cat. 26600.04) at 37 °C for 4 h. Cultures were centrifuged for 10 min at 4500 rpm, and pellets from 50 mL culture were resuspended in 5 mL sonication buffer; induced pellets containing TRX protein were resuspended in 5 M urea (Across Organics, Landsmeer, The Netherlands, Cat. 140750050), while Vim- and TRXtr-Vim containing pellets were resuspended in sonication buffer containing 2 M urea in PBS, 20% glycerol (VWR Chemicals, Amsterdam, The Netherlands, Cat. 24386.398), 0.1 µM EDTA (J.T.Baker, Deventer, The Netherlands, Cat. 1073), and 1% Triton X-100 (Amresco, Solon, OH, USA, Cat. M143). Bacterial suspensions were sonicated for 15 cycles (20 s on and 30 s off) with an amplitude of 22–24 microns using the Soniprep (150 MSE, London, UK). For Vim and TRXtr-Vim, 1 mM phenylmethylsulphonyl fluoride (PMSF, Sigma-Aldrich, Saint Louis, MO, USA, Cat. 93482) was added to the supernatant after sonication to prevent protein degradation. Finally, tubes were centrifuged at 4500rpm for 10 min, and the supernatant was stored at −20 °C until protein purification.
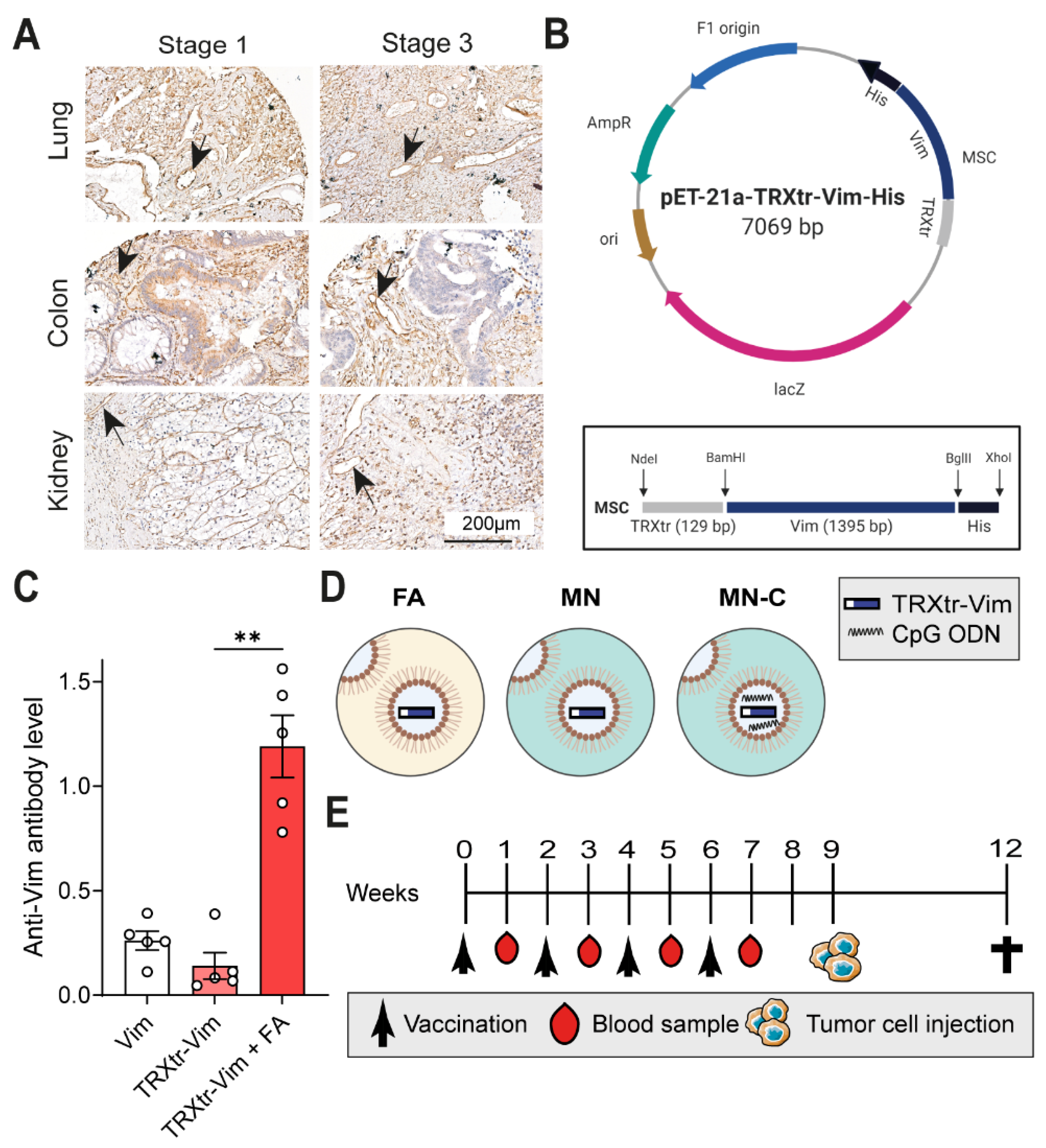
All three proteins contain a C-terminal His-tag (
Figure S1A–D), enabling purification using Ni-Affinity chromatography. Per 10 mL of thawed supernatants containing Vim or TRXtr-Vim, 300 µL Ni-NTA agarose slurry (Qiagen, Venlo, The Netherlands, Cat. 1018244) was added and incubated at 4 °C overnight. After centrifugation, the agarose beads were washed with wash buffer (PBS pH 7.0, 1M NaCl, 0.05% Tween-20) and transferred to a column with a glass filter (Sartorius Stedim Biotech, Göttingen, Germany, Cat. 82121-011-03). The column was washed with four 150µL fractions of 200 mM imidazole (J.T.Baker, Deventer, The Netherlands, Cat. 1747) and dissolved in 20 mM Tris pH 8.0 with 0.1 M NaCl, and the proteins were eluted with four 150 µL fractions of elution buffer (100 mM NaH
2PO
4 (Merck, Darmstadt, Germany, Cat. 1.06346.1000), 10 mM Tris·Cl, 8M urea, pH 4.5) (
Figure S1D). To purify the TRX protein, 10 mL of supernatant was incubated with 300 µL Ni-NTA agarose slurry in the presence of 10 mM imidazole at 4 °C overnight. The next day, the agarose beads were washed with 0.1 M NaCl, 0.1% Tween-20 in PBS and transferred to a column with a glass filter. The column was washed with 10 fractions of 20 mM imidazole in 20 mM Tris pH 8.0 with 0.1 M NaCl, and protein was eluted with four 150 µL fractions of 200 mM imidazole (
Figure S1D).
Protein content in elution fractions was visualized by SDS-PAGE using Mini-PROTEAN
® TGX 4–20% precast polyacrylamide gels (Bio-Rad, Veenendaal, The Netherlands, Cat. 4561094), stained with colloidal Coomassie brilliant blue G250 (Polysciences, Hirschberg an der Bergstrasse, Germany, Cat. 03707-50) (
Figure S1D). Fractions containing the protein of interest were dialyzed using 3.5 K SnakeSkin (Thermo Fisher Scientific, Landsmeer, The Netherlands, Cat. 68035) for TRX and 10K SnakeSkin (Thermo Fisher Scientific, Cat. 88245) for Vim and TRXtr-Vim. TRX protein fractions were directly dialyzed in PBS at 4 °C overnight. Vim and TRXtr-Vim proteins were dialyzed against 4 M urea in PBS at 4 °C overnight, followed by stepwise dialysis in 3 M (2 h), 2.5 M (2 h), and 2 M (2 h) urea. Final protein concentrations were determined with a Micro BCA Protein Assay (Thermo Fisher Scientific, Cat. 23235).
2.2. Animal Studies
All animal experiments were performed in accordance with Dutch guidelines and law on animal experimentation and were approved by the Centrale Commissie Dierproeven (CCD), registration no. DEC AngL13-02 and CCD AVD114002016576. Work protocols were approved by the VU-VUmc animal welfare body.
In the first in vivo experiment, three different experimental groups were compared, consisting of five 8-week-old female C57BL/6JOlaHsd mice (Envigo) each. All mice were vaccinated three times subcutaneously in the groin with an interval period of two weeks between immunizations. The first group was immunized with 30µg vimentin protein only and the second group with 40 µg of the conjugate vaccine TRXtr-Vim only. The final group was immunized with an emulsion containing 20 µg of TRXtr-Vim, mixed with Freund’s complete adjuvant (FCA, Sigma Aldrich, Zwijnsdrecht, The Netherlands, Cat. F-5881) in a 1:1 ratio. For the second and third vaccination, FCA was replaced by Freund’s incomplete adjuvant (FIA, Sigma Aldrich, Cat. F-5506). Blood samples were taken from the tail vein before the first vaccination and 1 week after each vaccination. Blood samples were coagulated overnight at 4 °C and centrifuged at 7000 rpm for 10 min at 4 °C in a microcentrifuge. The supernatant (serum) was removed, centrifuged again to remove additional debris, and stored at −20 °C until use.
Subsequently, the in vivo adjuvant comparison study was performed in duplicate and is indicated as ‘Study I’ and ‘Study II’ in other sections. Each treatment/control group contained ten 8-week-old female C57BL/6JOlaHsd mice (Envigo). Mice were immunized four times subcutaneously in the left groin with an interval period of two weeks between immunizations. Each vaccine emulsion contained 75 µg TRXtr-Vim or 10 µg TRX protein in a volume of 50 µL PBS (TRX) or 2 M urea (TRXtr-Vim), mixed with 50 µL adjuvant (ratio 1:1, aqueous phase: oil phase); total injection volume was 100 µL. In total, three different adjuvant compositions were compared. The first group received a primer vaccination with an emulsion containing FCA, followed by booster vaccinations with FIA. One study group received MN720 supplemented with 50 µg phosphorothioate CpG oligonucleotide 1826 (Eurogentec, Seraing, Belgium) per mouse during all 4 vaccinations, and the final group received MN720 alone, without any supplementation. All emulsions were mixed for 30 min on a Vortex-Genie 2 (Thermo Fisher Scientific) at full speed. Blood samples were taken from the tail vein at the start of the experiment before the first vaccination, one week after each vaccination, and at the end of the experiment. Blood samples were coagulated overnight at 4 °C, centrifuged, and the serum was stored at −20 °C until use.
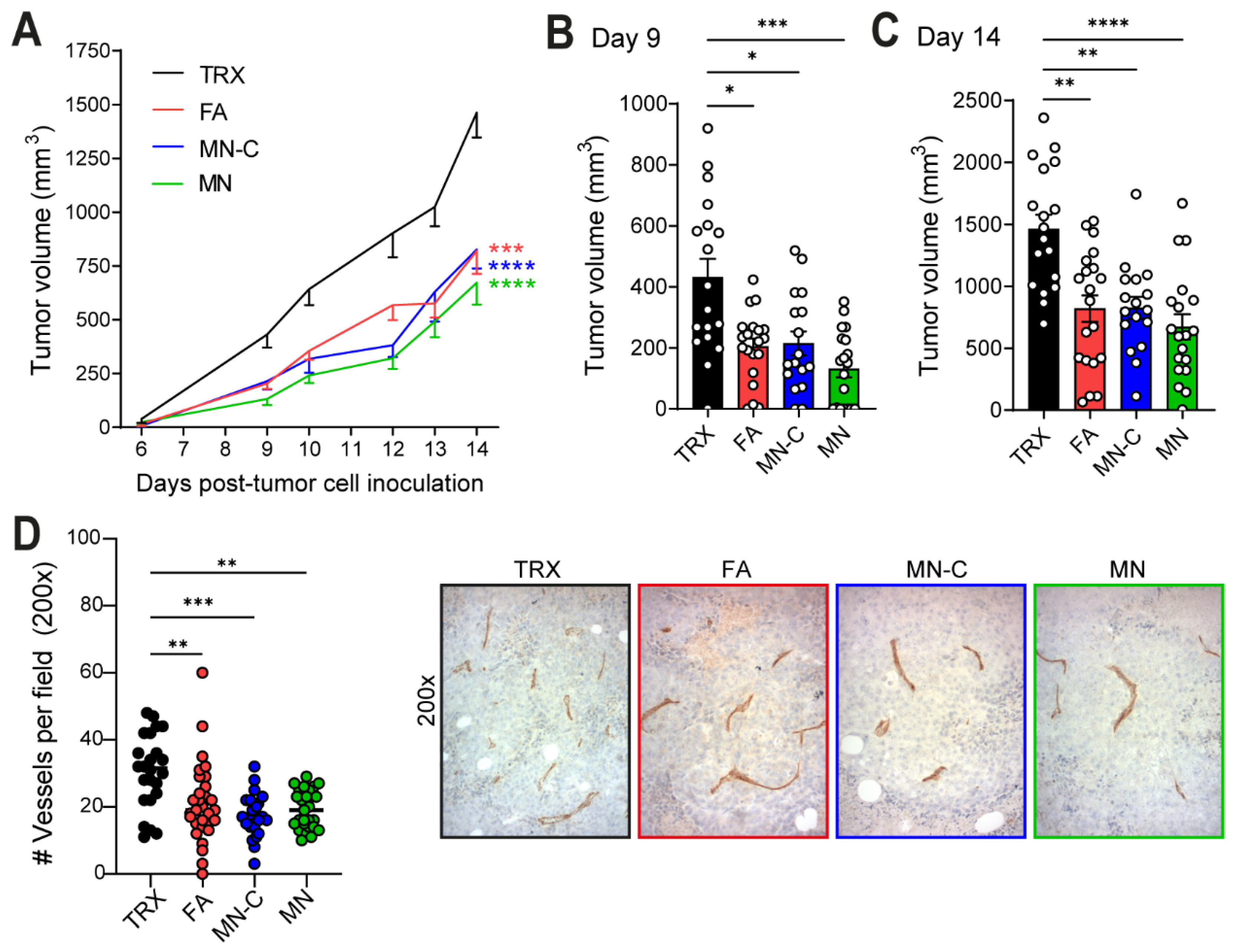
Murine B16F10 melanoma tumor cells (ATCC CRL-6475) were maintained in DMEM (Gibco, Waltham, MA, USA, Cat. 41965-039) supplemented with 1% of antibiotics (Penicillin/Streptomycin, Life Technologies, Carlsbad, CA, USA, Cat. 15140-122), 10% newborn calf serum (NBCS, Biowest, Nuaillé, France, Cat. S0750), and 2 mM L-glutamine (Roth, Karlsruhe, Germany Cat. HN08.2). One week after the last immunization, 1 × 105 B16F10 melanoma cells were inoculated subcutaneously in the left flank of C57BL/6 mice in a total volume of 100 μL (10% culture medium/PBS). Tumor growth was measured by calipers, and tumor volume was calculated with the formula: width2 × length × π/6. In Study I, two mice in the MN-C group and one mouse in the MN group showed no tumor take and were excluded from tumor growth data. Similarly, in Study II, one mouse in the MN-C group was excluded. In addition, two TRX control mice were sacrificed before tumor cell injection due to health issues. At the end of the experiment, mice were euthanized, and tumors and organs were removed and stored in 4% PFA/PBS (Electron Microscopy Sciences, Hatfield, PA, USA, Cat. 15710) overnight and consecutively paraffin-embedded.
2.3. Immunohistochemistry
Paraffin-embedded tumor tissues obtained during in vivo Study II were sectioned (3–5 µm) with a microtome (Leica, Nieuw-Vennep, The Netherlands). Sections were dried at least 24 h at 37 °C. Tumor necrosis was analyzed using hematoxylin and eosin (HE) staining. For further analyses regarding vessel density and immune cell content of the B16F10 tumor, tissue sections were used for immunohistochemistry. Before deparaffinization, tissue sections were incubated at 60 °C for 1 h, followed by 10 min at 56 °C on a heating plate. Slides were deparaffinized with xylene and consecutively rehydrated in 100%, 96%, and 70% ethanol and PBS. Endogenous peroxidase activity was blocked with a 15 min incubation in 0.3–3% hydrogen peroxidase in PBS (VWR Chemicals) at RT. Antigen retrieval was performed in citrate buffer (10 mM, pH 6.0) by autoclaving. For CD3 staining, TRIS/EDTA pH 9.0 buffer was used during antigen retrieval. Blocking was performed using 3% bovine serum albumin (BSA, Roche, Penzberg, Germany, Cat. 10735086001) for CD31, CD45, and CD11b or 20% horse serum (Sigma-Aldrich, Saint Louis, MO, USA, Cat. H1138) in PBS for vimentin for 1h at RT. A blocking solution of 4% BSA with 5% normal goat serum (Sigma-Aldrich, Saint Louis, MO, USA, Cat. G6767) was used for blocking in the CD3 staining procedure. For anti-vimentin staining on mouse sections, additional blocking with Fab fragments (Abcam, Cambridge, UK, Cat. ab6668) in a concentration of 50 µg/mL was performed for 2 h at RT.
Next, slides were incubated with the following primary antibodies: anti-CD31 rat anti-mouse IgG2a (1:50, Dianova, Hamburg, Germany, Cat. DIA-310M, clone SZ31), anti-CD45 rabbit anti-mouse (1:100, Abcam, Cat. Ab10558), anti-CD11b rabbit anti-mouse (1:4000, Abcam, Cat. Ab13357), anti-CD3 rabbit anti-mouse (1:600, Neomarkers, Fremond, CA, USA, Cat. RM-9717-S), and mouse anti-vimentin (Santa Cruz Biotechnology, Dallas, TX, USA, Cat. sc-373717, Clone E5, 1:50 dilution). After overnight incubation with primary antibodies at 4 °C, tissue sections were washed in PBS and incubated with biotinylated secondary antibodies for 45 min at RT. The following biotinylated secondary antibodies were used: swine anti-rabbit (1:500, Dako, Glostrup, Denmark, Cat. E0353), donkey anti-rat (1:500, Jackson lab, Baltimore, PA, USA, Cat. AB2340650), and goat anti-mouse (1:500, Dako, Cat. E0433). Slides were again washed in PBS and incubated with Strep-HRP (Dako, Cat. P0397) in 0.5% BSA for 30 min at RT. For CD3 staining, a goat anti-rabbit HRP polymer (Abcam, Cambridge, UK, Cat. Ab214880) was used. After washing with PBS, 3,3′-diaminobenzidine tetrahydrochloride hydrate (DAB, Sigma-Aldrich, Cat. D5637) staining was performed for up to 8 min at RT. Sections were washed with PBS and counterstained with Mayer’s hematoxylin (VWR Chemicals, Amsterdam, The Netherlands, Cat. 10047105) (1:4 diluted in 5 mM citrate buffer pH6.0) for 30 s. The reaction was stopped under running tap water for 10 min, and sections were dehydrated consecutively with 70%, 96%, and 100% ethanol. Finally, slides were incubated in xylene and mounted with Quick D mounting medium (Klinikpath, Duiven, The Netherlands, Cat. 7280) and covered with a coverslip. Sections were visualized with a 200× magnification on an Olympus BX50 microscope. Pictures were taken with a CMEX DC 5000C camera. The number of blood vessels or immune cells was counted per tissue core, with three fields in total per section. One tumor from the MN-C group and one tumor from the MN group were extensively necrotic and were excluded for CD31, CD45, CD11b, and CD3 quantification.
To validate vimentin expression in the tumor vasculature of human tumors, a tissue microarray was generated in collaboration with the biobank at the pathology department in Amsterdam UMC, location VUMC. The microarray contains tumors of stage 1 and stage 3, according to TNM classification. Tissue cores with a 1mm diameter were obtained from formalin-fixed and paraffin-embedded (FFPE) blocks using the TMA Grandmaster. The TMA tissue block was sectioned and stained using an anti-vimentin antibody (Santa Cruz Biotechnology, Dallas, TX, USA, Cat. sc-373717, clone E5, 1:50 dilution), as described above. No Fab fragments were needed during the staining procedure.
2.4. ELISA
Indirect ELISA was performed as described previously [
9]. Volumes per well were 50 µL, unless indicated otherwise. The 96-well ELISA plates (Thermo Fisher Scientific, Landsmeer, The Netherlands, Cat. 442404) were coated with 4 μg/mL recombinant vimentin (Vim) protein (see section protein production and purification) in 0.5 M urea in PBS for 1 h at 37 °C. To determine the antibody titers against TRX, ELISA plates were coated with TRX-gal1 according to Saupe et al. [
15]. Next, wells were blocked with 4% non-fat dry milk (Bio-Rad, Veenendaal, The Netherlands, Cat. 1706404) in PBS for 1h at 37 °C using 100 µL per well. Plates were washed one time with PBS for 1 min to remove the excess blocking solution.
Mouse sera were briefly centrifuged and diluted 1:10 in 4% non-fat dry milk in PBS. For TRX antibody titers, 5 mouse sera were pooled per measurement to reduce the amount of serum needed. Sera were further diluted in Rosetta Gami extract, which is used to reduce the non-specific binding of serum antibodies to the recombinantly produced vimentin coating. Rosetta Gami extract was prepared by overnight culturing of Rosetta Gami DE3 pET21a-TRX bacteria at 37 °C at 200 rpm. The next day, the culture was centrifuged for 20 min at 4500 rpm, and the culture supernatant was discarded. Bacterial pellets were resuspended in PBS and sonicated for 15 cycles (20 s on and 30 s off) with an amplitude of 22–24 microns. After sonication, tubes were centrifuged at 4500 rpm for 10 min, and the supernatant was stored at −20 °C to be used as Rosetta Gami extract during ELISA.
Diluted sera were incubated in the 96-well plate for 45 min at 37 °C, followed by four washing steps with PBS. After washing, plates were incubated with biotinylated polyclonal goat anti-mouse Ig (Dako Cytomation, Glostrup, Denmark, Cat. E0433) and diluted 1:2000 in 0.01% PBS-T for 45 min at 37 °C, followed by four washing steps with PBS. Thereafter, wells were incubated with streptavidin–horseradish peroxidase (Dako Cytomation, Glostrup, Denmark, Cat. P0397) for 30 min at 37 °C, which was also diluted 1:2000 in 0.01% PBS-T. After four washes with PBS, wells were incubated with TMB substrate (Sigma-Aldrich, Saint Louis, MO, USA, Cat. T-8665) for 5 min, and absorbance was measured at 655 nm using a Biotek Synergy HT microplate reader (Biotek, Bad Friedrichshall, Germany). Antibody titers were determined by calculating at which serum dilution the titration curve would give an OD value of 0.2. This threshold was set based on the background signal observed in blank control wells and wells incubated with serum retrieved from mice vaccinated with the control TRX vaccine.
To determine the antibody isotypes of the anti-vimentin antibodies in mouse sera, a similar ELISA protocol was used as described above. However, for each measurement, sera of five different mice were pooled, and blocking was performed using 3% bovine serum albumin (BSA, Roche, Penzberg, Germany, Cat. 10735086001). In addition, the following isotype-specific biotinylated goat anti-mouse secondary antibodies were used: IgG1 (Southern Biotech, Birmingham, AL, USA, Cat. 1070 08), IgG2b (Southern Biotech, Birmingham, AL, USA, Cat. 1090 08), IgG2c (Southern Biotech, Birmingham, AL, USA, Cat, 1079 08), IgG3 (Southern Biotech, Birmingham, AL, USA, Cat. 1100 08), IgM (Southern Biotech, Birmingham, AL, USA, Cat. 1020 08), and IgA (Southern Biotech, Birmingham, AL, USA, Cat. 1040 08). Listed antibodies were diluted 1:2500 in 0.01% PBS-T and incubated for 45 min at 37 °C.
2.5. Surface Plasmon Resonance Using Biacore
All the surface plasmon resonance (SPR) experiments were done using Biacore T200 (Cytiva). Recombinant murine Vim protein was immobilized on the CM5 chip (Cytiva, Freiburg im Breisgau, Germany) surface using amine coupling at pH 4.0. All the serum samples obtained from Study I were injected over the measurement and reference cells at a flow rate of 20 μL/min for 120 sec in 2 mM Na Phosphate buffer pH 7.5, 0.05% Tween-20. To normalize the anti-vimentin antibody input from all sera, the serum dilution was normalized based on the determined anti-vimentin antibody titer by ELISA. At the end of each measurement cycle, the surface was regenerated by subsequent injections of 10 mM Glycine pH 1.5 and 0.5 M urea. Injection of a commercial anti-mouse vimentin antibody (Clone E5, Santa Cruz Biotechnology, Dallas, TX, USA, Cat. sc-373717) was used as the positive control. All samples were run in quadruplicate, and for each group, one mouse was excluded due to high variation between the technical replicates. Initial data analysis was done using Biacore T200 Evaluation Software v 3.2 (Cytiva, Freiburg im Breisgau, Germany).
2.6. LEGENDplex
The LEGENDplex™ MU Th Cytokine Panel (12-plex) w/ VbP V03 (BioLegend, London, United Kingdom, Cat. 741044) was used for quantification of cytokines in mouse sera, collected one week after the fourth vaccination from both Studies I and II. For FA, three out of 20 mice did not have a sufficient amount of serum for analysis, as well as one mouse from the MN-C-vaccinated group. Measurements were performed according to the manufacturer’s protocol and were read on a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed using LEGENDplex™ Data Analysis Software Suite.
2.7. Splenocyte Isolation, Restimulation, and FACS Analysis
Spleens from mice immunized with TRX or TRXtr-Vim in combination with Freund’s adjuvant were isolated and cut into small pieces and mechanically dissociated. Splenocytes were passed through a 70 μm cell strainer (Corning, Amsterdam, The Netherlands, Cat. 431751) and centrifuged for 5 min at 1500 rpm. Red blood cells were lysed using a buffer containing 150 mM NH4Cl, 10 mM KHCO3, and 100 mM EDTA (pH = 7.4) for 3 min. Splenocytes were washed and resuspended in RPMI-1640 (Biowest, Nuaillé, France, Cat. L0495-500) medium supplemented with 1% of antibiotics (Penicillin/Streptomycin, Life Technologies, Carlsbad, CA, USA, Cat. 15140-122), 10% newborn calf serum (NBCS, Biowest, Cat. S0750), 2 mM L-glutamine (Roth, Karlsruhe, Germany, Cat. HN08.2), and 50 μM 2-mercaptoethanol (Gibco, Waltham, MA, USA, Cat. 31350-010). Isolated splenocytes were seeded in U-bottom 96-well plates with 2×106 cells/well and were restimulated ex vivo for 48 h with or without recombinant endotoxin free vimentin (see section vectors and vaccine production) at a concentration of 20 µg/mL. Endotoxin was removed while the vimentin protein was bound to the Ni-agarose by washing the column with 20–50 column volumes of PBS-0.1% Triton X-114 (Sigma-Aldrich, Zwijnsdrecht, The Netherlands, Cat. X114-100 mL) at 4 °C in the cold room. Thereafter, the column was washed with 5–20 column volumes of PBS at 4 °C to remove the Triton X-114. Endotoxin levels were determined by LAL-assay (Thermo Scientific, Landsmeer, The Netherlands, Pierce Chromogenic Endotoxin Quant kit, Cat. A39552S). During the final 6 h of stimulation, Brefeldin A (BioLegend, London, United Kingdom, Cat. 420601) was added to all wells. After restimulation, cells were transferred to a V-bottom 96-well plate and washed with PBS. Cells were resuspended in Fc blocking solution (BioLegend, London, United Kingdom, Cat. 101320) for 10 min at room temperature. Next, cells were incubated with anti-mouse CD4-AF700 (1:200, BioLegend, London, United Kingdom, Cat. 100429), anti-mouse CD8b-PE (1:200, BioLegend, Cat. 126627), and Zombie Aqua fixable viability dye (1:200, BioLegend, London, United Kingdom, Cat. 423101) diluted in PBS for 20 min on ice. Cells were washed with PBS and fixed with 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Cat. 15710) for 15 min on ice. After washing, cells were permeabilized using an intracellular staining permeabilization wash buffer (BioLegend, London, United Kingdom, Cat. 421002). Cell suspensions were then incubated with anti-mouse IFNγ-APC (1:200, BioLegend, London, United Kingdom, Cat. 505809) and TNFα-PE (1:200, BioLegend, London, United Kingdom, Cat. 506305) diluted in permeabilization buffer for 30 min at room temperature. Cells were washed twice with the permeabilization buffer and finally resuspended in 100 μL PBS. Samples were measured using the LSRII Fortessa (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer. Data analysis was performed with the FlowJo V10.8.1 software (Tree Star).
2.8. Statistics
All statistical tests were executed using GraphPad Prism 9.1.0 (GraphPad Software Inc., La Jolla, CA, USA), and * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001 were considered statistically significant. Error bars represent the standard error of the mean (SEM) unless specified otherwise. For comparison of tumor growth curves and antibody isotype distributions, a two-way ANOVA was used. Other comparisons for different treatment groups were tested using a Kruskal–Wallis test with Dunn’s multiple comparison testing. Correlation plots were tested for statistical significance using simple linear regression analysis.
4. Discussion
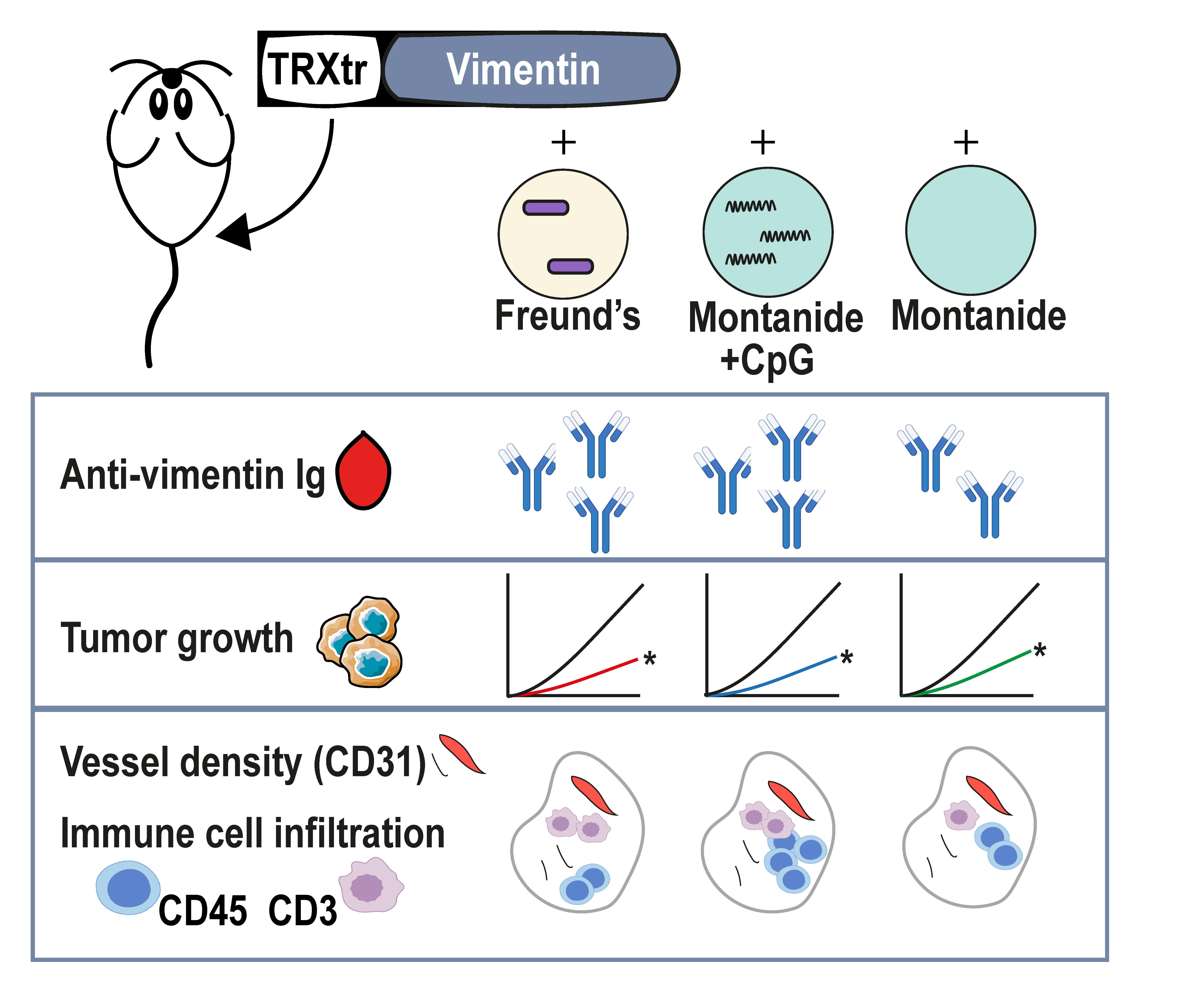
We have recently shown that extracellular vimentin is overexpressed and secreted by tumor endothelial cells and that anti-vimentin antibodies are capable to inhibit tumor growth in several in vivo murine tumor models [
9]. It was demonstrated that vaccination with the iBoost conjugate TRXtr-Vim vaccine generates a potent humoral immune response when coadministered with the gold standard Freund’s adjuvant (FA) (
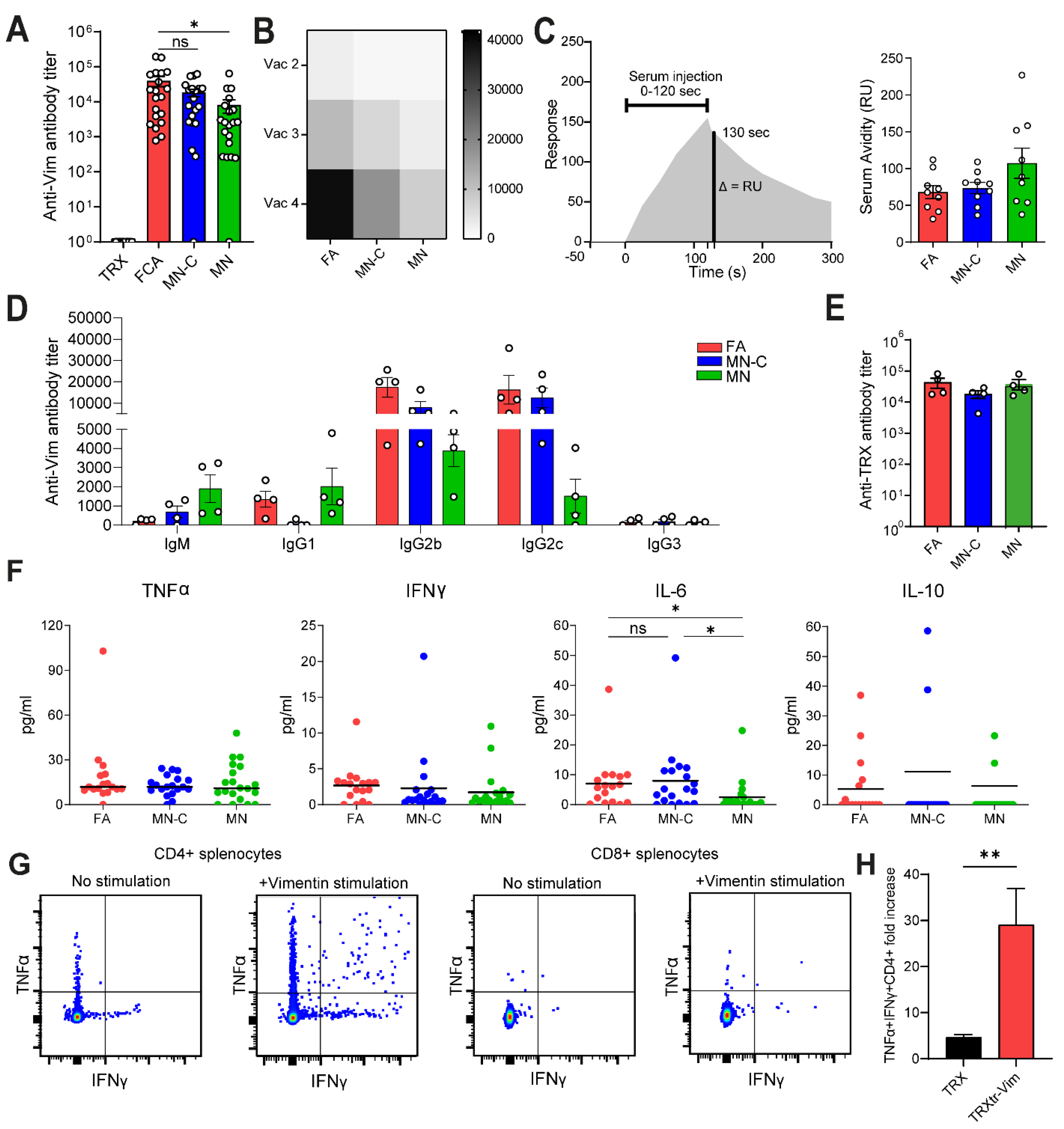
Figure 1C). Although FA is highly immune stimulatory, it is not approved for clinical use due to its toxicity. Therefore, there is a high need to identify a non-toxic alternative that generates a comparable immune response, leading to efficient anti-cancer activity. In the current study, we observed that the formulation of Montanide ISA 720 supplemented with CpG oligodeoxynucleotide 1826, but not Montanide alone, results in a similar humoral immune response towards vimentin, as compared to FA. We detected comparable anti-vimentin antibody titers in the serum of vaccinated mice (
Figure 2A) and could confirm that both adjuvants resulted in the generation of IgG2b and IgG2c antibodies. Since these isotypes can bind to all four types of FcγR types, they play a major role in antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cellular cytotoxicity (ADCC) [
19].
To investigate whether the addition of CpG was necessary for a potent anti-vimentin immune response, we included one experimental group in which mice were vaccinated with TRXtr-Vim and Montanide adjuvant (MN) and one group where the adjuvant was supplemented with CpG (MN-C). Unmethylated CpG motifs are abundantly present in prokaryotic DNA, which is released during bacterial infections [
21], generating strong immune stimulation. These unmethylated CpG motifs can bind to TLR9 and activate several pathways such as IFN type I signaling [
15]. In mice, the TLR9 is expressed by monocytes, macrophages, and conventional dendritic cells [
21], while in humans mainly B cells and plasmacytoid dendritic cells express the receptor [
22]. We observed that vaccination with MN alone resulted in lower IL-6 production (
Figure 2F), lower immune cell infiltration (
Figure 4A), and a lower percentage of IgG2c antibodies (
Figure S2C) compared to MN-C vaccination. Interestingly, IL-6 has been described to have an important role in the differentiation of B cells into antibody-producing plasma cells, a crucial step in our vaccination strategy [
23]. Together these data indicated that CpG addition has a beneficial effect on the immune response following TRXtr-Vim vaccination. This effect might partly be explained by an enhanced maturation and activation of the dendritic cells that capture the protein vaccine after injection. Our data are in accordance with a paper by Johansson et al., in which several commonly used adjuvants were compared in their potential to break self-tolerance against immunoglobulin E (IgE) [
24]. They showed that vaccination with the well-known adjuvant Alum did not induce antibodies against the self-antigen IgE. However, they did detect anti-IgE antibodies in the serum of rats vaccinated with the biodegradable adjuvant Montanide ISA 720. Furthermore, Huijbers et al. previously showed that Montanide ISA 720, supplemented with CpG, was also successful in the induction of antibodies against ED-B [
13,
15] and ED-A [
8].
In addition to CpG, there are many more immunostimulatory compounds that can be used during vaccination. For example, Freund’s complete adjuvant is complemented with heat-killed and dried Mycobacterium tuberculosis (MT). Mycobacteria ligands can bind a broad range of receptors such as TLR2, TLR4, TLR8, and TLR9 [
18,
25]. When characterizing the humoral response after vaccination with Montanide supplemented with MT, antibody titers against vimentin were comparable to MN-C-vaccinated mice (data not shown). Since MT was not superior to CpG and no clinical data is yet available on Montanide ISA 720 supplemented with MT, we did not continue with this adjuvant composition in subsequent experiments. Ringvall et al. also directly compared the potency of CpG, double-stranded RNA polyC:poly G (TLR3 ligand), and muramyl dipeptide (NOD2 ligand) [
26], three commonly used compounds in vaccine adjuvants. They showed that vaccination with Montanide supplemented with CpG resulted in the highest level of anti-self IgE antibodies, with levels comparable to FA.
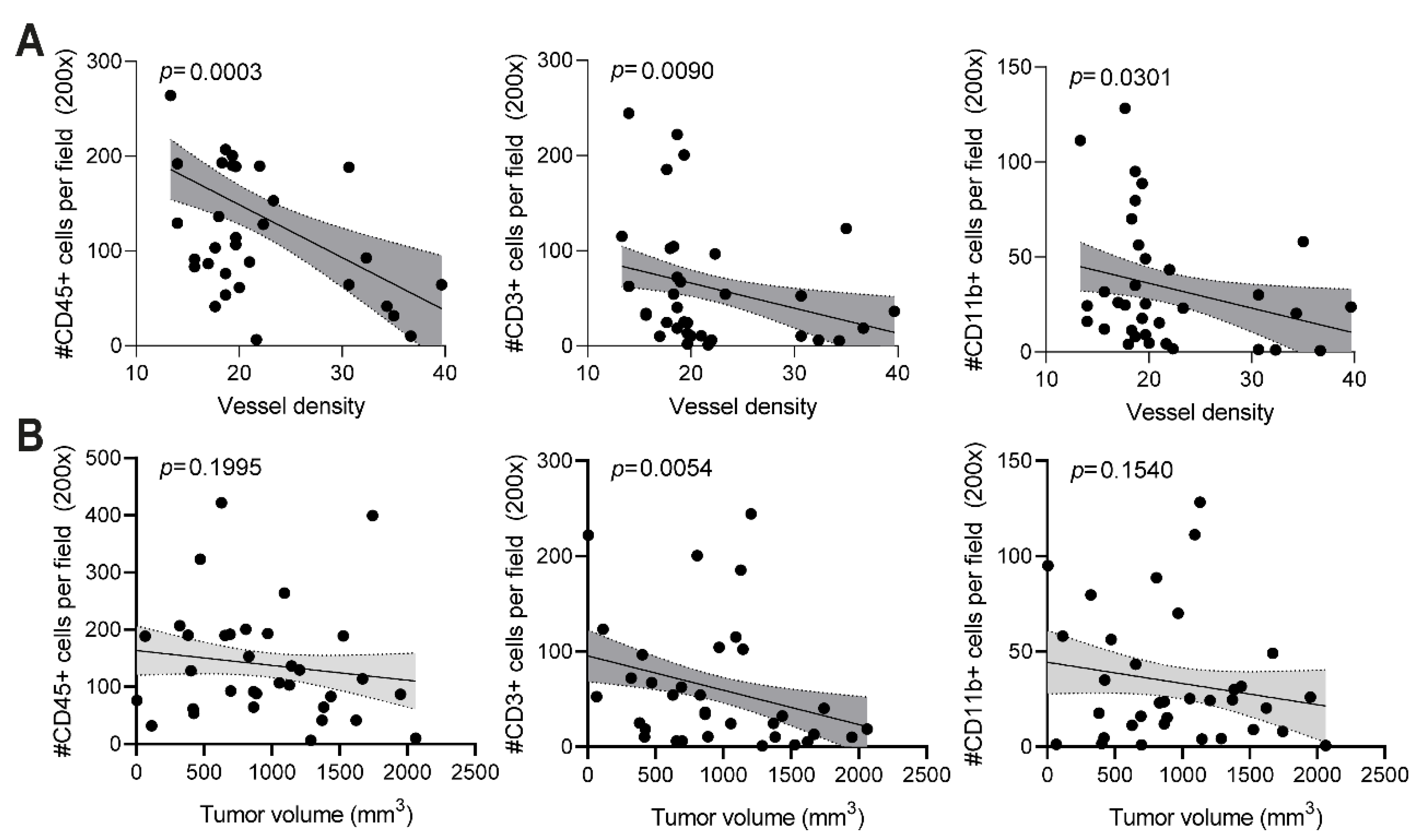
The anti-vimentin antibodies that are generated as a result of vaccination with a potent adjuvant are thought to play a major role in the observed reduced vessel density and anti-tumor effect. When anti-vimentin antibodies bind their target in the tumor vasculature, the Fc region of the antibody can bind to an activating FcγR on the surface of several types of immune cells, such as macrophages, neutrophils, and NK cells. The activation of immune cells can trigger effector functions, which eventually leads to tumor vessel destruction, as previously suggested for ED-B [
11]. When quantifying the number of blood vessels in B16F10 tumor sections of TRXtr-Vim-vaccinated mice, we indeed found a significantly decreased vessel density (
Figure 3D). Interestingly, a decreased vessel density was negatively correlated with an enhanced number of CD45+ immune cells in the tumor (
Figure 5A). Increased infiltration of immune cells after vimentin vaccination has recently also been shown by van Beijnum et al. [
9] and can potentially be explained by several theories. Firstly, vimentin vaccination upregulates intercellular adhesion molecule (ICAM)-1 expression in the tumor vasculature, which can bind to the leukocyte function-associated antigen (LFA)-1 integrin on leukocytes, facilitating their transmigration into the tumor. Secondly, the P-selectin glycoprotein ligand 1 (PSGL-1) on leukocytes is also involved in binding to the endothelium by binding to its receptor P-selectin. Lam et al. showed that neutrophil adhesion to endothelial cells is reduced in the presence of vimentin, an observation they linked to direct interaction between vimentin and P-selectin [
27].
In addition to the total number of immune cells, we also quantified the number of CD3+ T cells in the B16F10 tumors by immunohistochemistry (
Figure 4C,D). Vaccination with FA resulted in the strongest T cell infiltration, followed by MN-C. In addition to the upregulation of ICAM-1, it has been shown that TRXtr-Vim vaccination can downregulate vascular PD-L1 expression in B16F10 tumors [
9]. Since PD-L1 is involved in T cell exhaustion by interacting with PD-1 on T cells, vimentin vaccination can enhance the fitness of the infiltrating T cells. Interestingly, the number of T cells showed a significant negative correlation with both the vessel density (
Figure 5A) and final tumor volume (
Figure 5B). Based on CD3 expression, we were unable to differentiate between T helper cells (CD3+CD4+) and cytotoxic T lymphocytes (CD3+CD8+). Flow cytometry analysis on B16F10 tumors after TRXtr-Vim vaccination with Freund’s adjuvant by van Beijnum et al. [
9] showed that CD4+ T cells account for approximately 25% of total intratumoral immune cells, while 15% consists of CD8+ cytotoxic T cells. This ratio between CD4+ and CD8+ T cells is similar in tumors after TRXtr-Vim and TRX vaccination. However, the general increase in CD3+ T cells after TRXtr-Vim vaccination (
Figure 4C), leads to the hypothesis that both subsets are enriched in the tumor microenvironment after vimentin vaccination. T helper cells can, in addition to their important role in activating vimentin-specific plasma cells, secrete pro-inflammatory cytokines such as IFNγ and TNFα [
28].
Ex vivo stimulation of splenocytes from TRXtr-Vim-vaccinated mice with recombinant vimentin protein showed that no vimentin-specific cytotoxic T cells were induced by vimentin vaccination (
Figure 2G,H). However, cytotoxic T cells can still play a major role by attacking tumor cells directly. The enhanced infiltration of T cells after TRXtr-Vim vaccination shows potential for several types of combination therapies, such as checkpoint inhibition or Chimeric Antigen Receptor (CAR)-T cells [
29].
The quantification of the number of CD11b+ immune cells showed that the highest infiltration of myeloid cells was observed in mice vaccinated with MN-C and MN (
Figure 4E,F). This CD11b+ immune cell population can contain a large number of different myeloid immune cell subsets, such as monocytes, macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs). Also NK cells can express CD11b on their cell surface [
30]. Based on previous experiments performed by van Beijnum et al., we know that TRXtr-Vim vaccination can enhance the infiltration of several anti-tumor immune subsets such as DCs and NK cells. Importantly, we also observed a reduction in the immunosuppressive MDSCs in the tumors from TRXtr-Vim-vaccinated mice [
9]. Since these MDSCs are known to hinder the anti-tumor effects of NK cells, B cells, and T cells, a reduction in this immune subset shows that TRXtr-Vim vaccination generates a less immunosuppressive tumor microenvironment [
31]. However, further studies are needed to pinpoint which of these subsets is most abundantly present in the tumor tissues after vaccination with Montanide adjuvant.
Finally, it is important to mention that the Montanide ISA 720 adjuvant has already been tested in several clinical trials. Vaccination against a malaria antigen was safe and well-tolerated, with only mild local reactions at the injection site [
32]. Montanide ISA 720 has also already been investigated in several cancer vaccines, such as peptide-based vaccines for melanoma with [
33] or without the addition of human CpG 7909 [
34]. In these melanoma patients, redness at the injection sites was observed. In addition, two patients showed extensive redness and swelling over their lower abdomen, which resolved within two weeks after immunization [
34]. In the current murine studies, we did not observe signs of toxicity associated with the Montanide adjuvant, with or without the addition of CpG, indicating that the adjuvant is also well-tolerated in combination with the TRXtr-Vim vaccine. In addition, we previously extensively studied the potential toxicity of vimentin vaccination in both mice and dogs [
9]. We did not observe significant weight loss in mice after TRXtr-Vim vaccination, even when kept hyperimmune for up to a year. Secondly, the TRXtr-Vim vaccine had no effect on wound healing. Finally, the vaccine is well-tolerated in a clinical study in client-owned dogs with spontaneous bladder cancer. We did not observe systemic adverse events, but there were local mild reactions at the injection site [
9].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




