Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Animals and Tumor Models
2.4. Uptake and Binding of FOSCAN in MC-38 and CT-26 Cells
2.5. Intracellular Fluorescence Imaging of FOSCAN in MC-38 and CT-26 Cells
2.6. PDT Effects of FOSCAN In Vitro
2.7. ATP Release by FOSCAN-PDT-Treated Tumor Cells In Vitro
2.8. Flow Cytometry Analysis of FOSCAN-PDT-Treated Tumor Cells In Vitro
2.9. Phagocytosis of DCs
2.10. Dendritic Cell Activation and Maturation Induced by FOSCAN-PDT-Treated Tumor Cells In Vitro
2.11. Characteristics of HBc VLPs In Vitro
2.12. DC and B Cell Activation Study by HBc VLPs In Vitro
2.13. FOSCAN-Mediated PDT in Combination with Therapeutic HBc VLP Treatments In Vivo
2.14. Analysis of Spleen-Derived Immune Cell Populations after Treatments
2.15. Blood Analysis for Immune RESPONSES after treatments
2.16. Tumor Re-Challenge and Immune Memory Analysis
2.17. Statistical Analysis
3. Results
3.1. FOSCAN Uptake, Binding, and Localization In Vitro
3.2. Tumor Cell Responses to PDT In Vitro
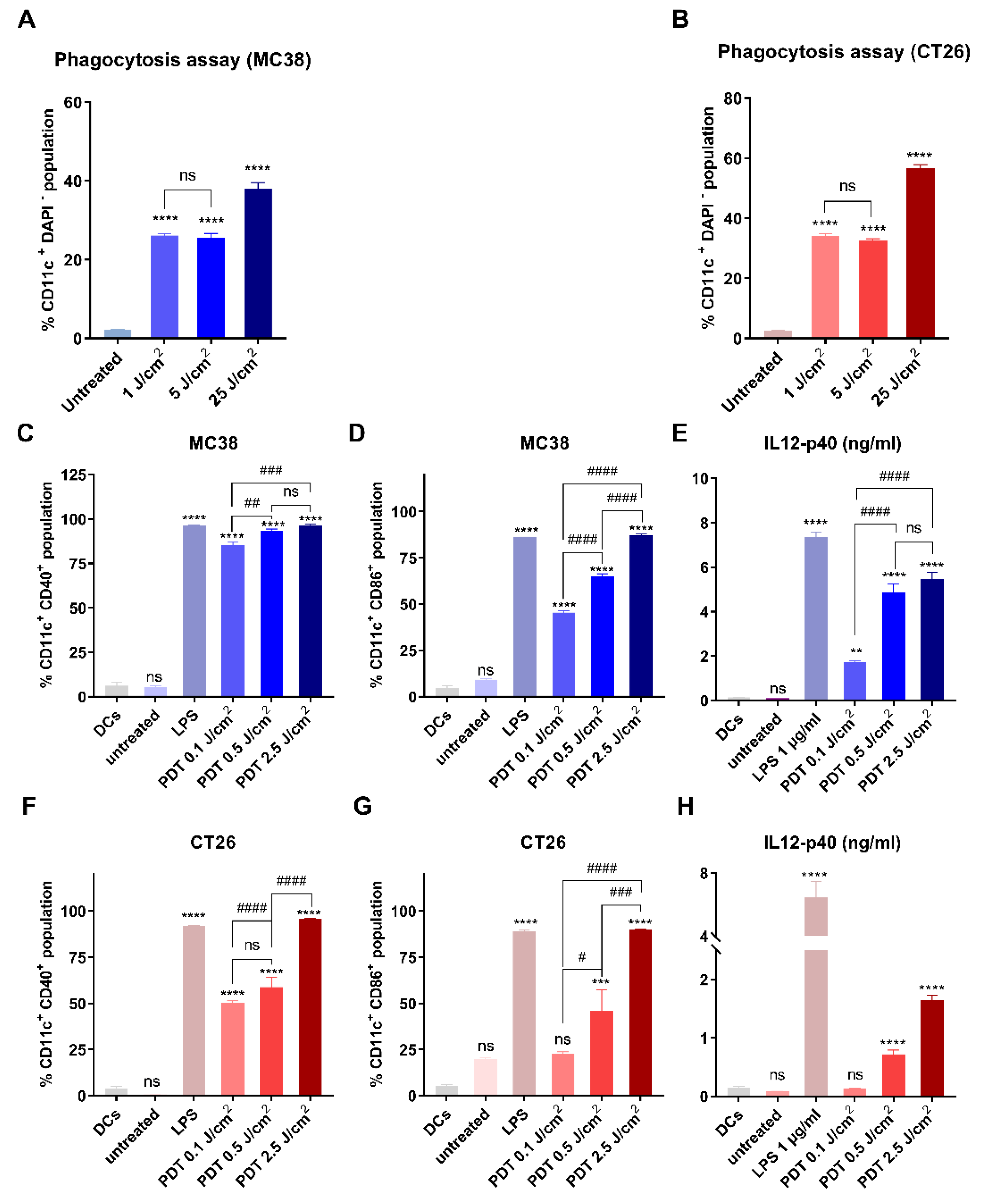
3.3. Immunological Effects of FOSCAN-PDT-Treated Cancer Cells
3.4. Physical and Biological Properties of HBc VLPs In Vitro
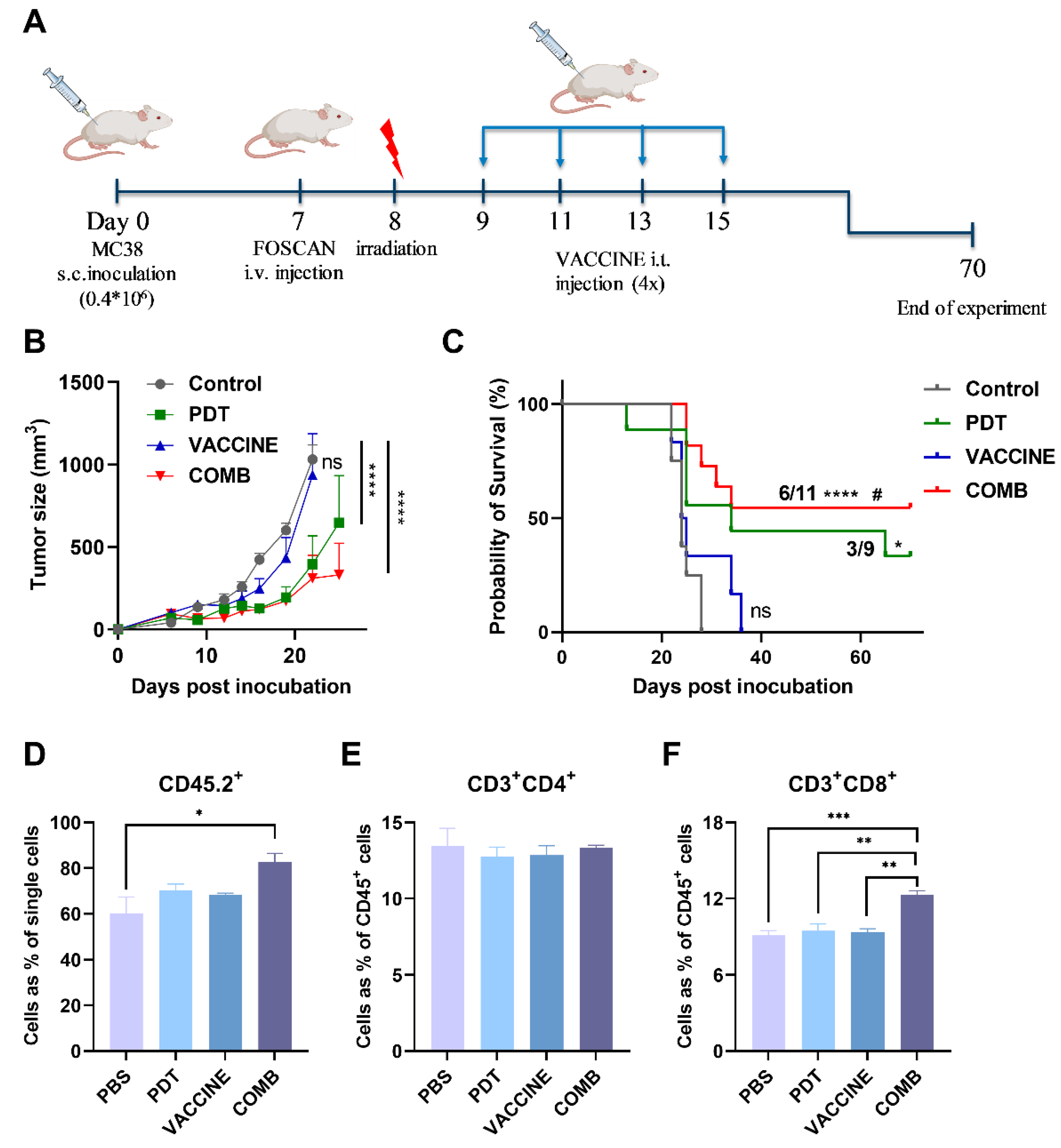
3.5. The Combination of FOSCAN-PDT Together with HBc VLPs Vaccine Inhibits Tumor Growth and Increases the Survival Rate of Tumor-Bearing Mice
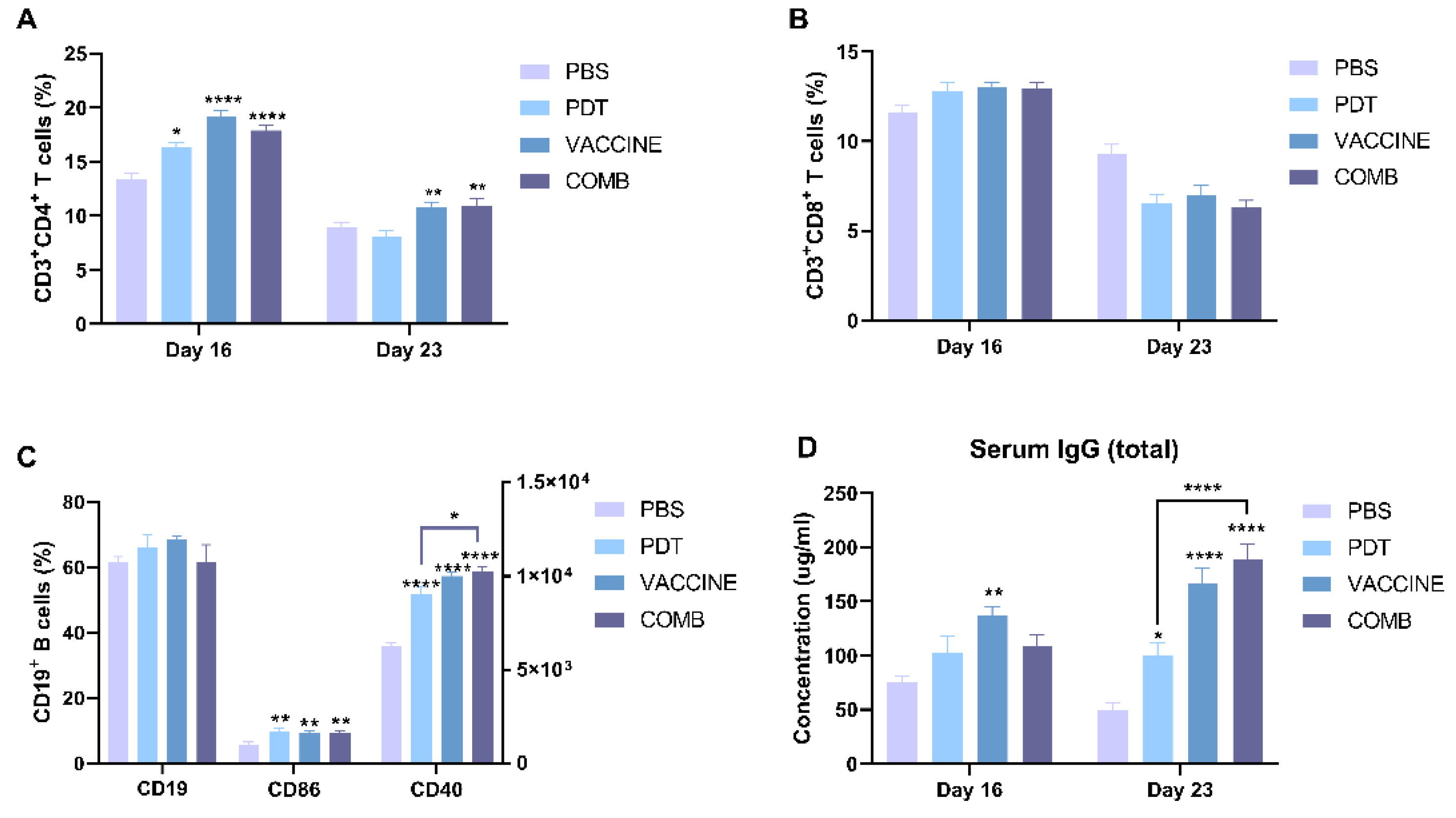
3.6. The Combination of FOSCAN-PDT Together with the HBc VLP Vaccine Induces Antitumor Humoral Immunity
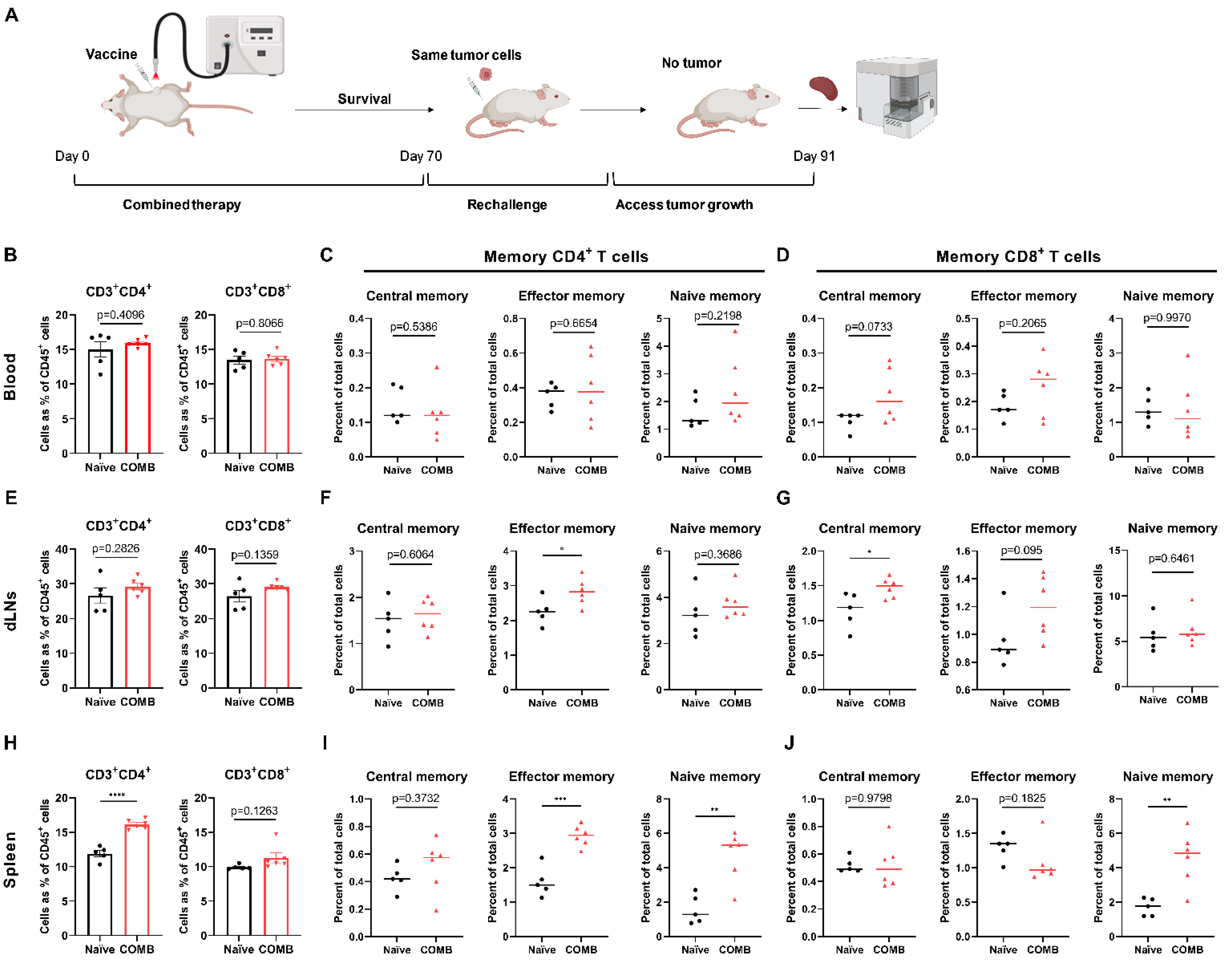
3.7. The Combination of FOSCAN-PDT Together with HBc VLP Vaccine Modulates the Function of Immune Memory Cells to Protect MICE from Tumor Rechallenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in CA Cancer J. Clin. 2020, 70, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M. Colon Cancer: A Clinician’s Perspective in 2019. Gastroenterol. Res. 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, K.; Ding, L.; Yan, H.; Hba, M.E.; Chadi, S.; Quereshy, F.; Zheng, G. Photodynamic Therapy for Colorectal Cancer: A Systematic Review of Clinical Research. Surg. Innov. 2022, 15533506221083545. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Allison, R.R.; Moghissi, K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin. Endosc. 2013, 46, 24–29. [Google Scholar] [CrossRef]
- Michels, S.; Schmidt-Erfurth, U. Sequence of early vascular events after photodynamic therapy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2147–2154. [Google Scholar] [CrossRef] [Green Version]
- Kawczyk-Krupka, A.; Bugaj, A.M.; Latos, W.; Zaremba, K.; Wawrzyniec, K.; Kucharzewski, M.; Sieroń, A. Photodynamic therapy in colorectal cancer treatment--The state of the art in preclinical research. Photodiagnosis Photodyn. Ther. 2016, 13, 158–174. [Google Scholar] [CrossRef]
- Cramers, P.; Ruevekamp, M.; Oppelaar, H.; Dalesio, O.; Baas, P.; Stewart, F.A. Foscan uptake and tissue distribution in relation to photodynamic efficacy. Br. J. Cancer 2003, 88, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Story, W.; Sultan, A.A.; Bottini, G.; Vaz, F.; Lee, G.; Hopper, C. Strategies of airway management for head and neck photo-dynamic therapy. Lasers Surg. Med. 2013, 45, 370–376. [Google Scholar] [CrossRef]
- Bown, S.G.; Rogowska, A.Z.; Whitelaw, D.E.; Lees, W.R.; Lovat, L.B.; Ripley, P.; Jones, L.; Wyld, P.; Gillams, A.; Hatfield, A.W.R. Photodynamic therapy for cancer of the pancreas. Gut 2002, 50, 549–557. [Google Scholar] [CrossRef]
- Pereira, S.P.; Ayaru, L.; Rogowska, A.; Mosse, A.; Hatfield, A.R.; Bown, S.G. Photodynamic therapy of malignant biliary strictures using meso-tetrahydroxyphenylchlorin. Eur. J. Gastroenterol. Hepatol. 2007, 19, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Wyss, P.; Schwarz, V.; Dobler-Girdziunaite, D.; Hornung, R.; Walt, H.; Degen, A.; Fehr, M. Photodynamic therapy of locoregional breast cancer recurrences using a chlorin-type photosensitizer. Int. J. Cancer 2001, 93, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Huis In’t Veld, R.V.; Ritsma, L.; Kleinovink, J.W.; Que, I.; Ossendorp, F.; Cruz, L.J. Photodynamic cancer therapy enhances the accumulation of nanoparticles in tumor-associated myeloid cells. J. Control. Release 2020, 320, 19–31. [Google Scholar] [CrossRef] [PubMed]
- van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Hao, Y.; Chung, C.K.; Yu, Z.; Huis in ‘t Veld, R.V.; Ossendorp, F.A.; ten Dijke, P.; Cruz, L.J. Combinatorial Therapeutic Approaches with Nanomaterial-Based Photodynamic Cancer Therapy. Pharmaceutics 2022, 14, 120. [Google Scholar] [CrossRef]
- Fleites, Y.A.; Aguiar, J.; Cinza, Z.; Bequet, M.; Marrero, E.; Vizcaíno, M.; Esquivel, I.; Diaz, M.; Sin-Mayor, A.; Garcia, M.; et al. HeberNasvac, a Therapeutic Vaccine for Chronic Hepatitis B, Stimulates Local and Systemic Markers of Innate Immunity: Potential Use in SARS-CoV-2 Postexposure Prophylaxis. Euroasian J. Hepatogastroenterol. 2021, 11, 59–70. [Google Scholar] [CrossRef]
- Moradi Vahdat, M.; Hemmati, F.; Ghorbani, A.; Rutkowska, D.; Afsharifar, A.; Eskandari, M.H.; Rezaei, N.; Niazi, A. Hepatitis B core-based virus-like particles: A platform for vaccine development in plants. Biotechnol. Rep. 2021, 29, e00605. [Google Scholar] [CrossRef]
- Zhang, E.; Lu, M. Toll-like receptor (TLR)-mediated innate immune responses in the control of hepatitis B virus (HBV) infection. Med. Microbiol. Immunol. 2015, 204, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.; Storni, T.; Manolova, V.; Didierlaurent, A.; Sirard, J.-C.; Röthlisberger, P.; Bachmann, M.F. Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur. J. Immunol. 2003, 33, 1465–1470. [Google Scholar] [CrossRef]
- Al Mahtab, M.; Akbar, S.M.F.; Aguilar, J.C.; Guillen, G.; Penton, E.; Tuero, A.; Yoshida, O.; Hiasa, Y.; Onji, M. Treatment of chronic hepatitis B naïve patients with a therapeutic vaccine containing HBs and HBc antigens (a randomized, open and treatment controlled phase III clinical trial). PLoS ONE 2018, 13, e0201236. [Google Scholar] [CrossRef]
- Lazdina, U.; Cao, T.; Steinbergs, J.; Alheim, M.; Pumpens, P.; Peterson, D.L.; Milich, D.R.; Leroux-Roels, G.; Sällberg, M. Molecular basis for the interaction of the hepatitis B virus core antigen with the surface immunoglobulin receptor on naive B cells. J. Virol. 2001, 75, 6367–6374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, P.; Stober, D.; Oehninger, C.; Melber, K.; Reimann, J.; Schirmbeck, R. Priming Th1 immunity to viral core particles is facilitated by trace amounts of RNA bound to its arginine-rich domain. J. Immunol. 2002, 168, 4951–4959. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Tal, G.; Lider, O.; Shaul, Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J. Immunol. 2005, 175, 3165–3176. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.O.; Tucker, A.; Frelin, L.; Sallberg, M.; Jones, J.; Peters, C.; Hughes, J.; Whitacre, D.; Darsow, B.; Peterson, D.L.; et al. Interaction of the hepatitis B core antigen and the innate immune system. J. Immunol. 2009, 182, 6670–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mahtab, M.; Akbar, S.M.; Aguilar, J.C.; Uddin, M.H.; Khan, M.S.; Rahman, S. Therapeutic potential of a combined hepatitis B virus surface and core antigen vaccine in patients with chronic hepatitis B. Hepatol. Int. 2013, 7, 981–989. [Google Scholar] [CrossRef]
- Riedl, P.; Buschle, M.; Reimann, J.; Schirmbeck, R. Binding immune-stimulating oligonucleotides to cationic peptides from viral core antigen enhances their potency as adjuvants. Eur. J. Immunol. 2002, 32, 1709–1716. [Google Scholar] [CrossRef]
- Akbar, S.M.; Yoshida, O.; Chen, S.; Cesar, A.J.; Abe, M.; Matsuura, B.; Hiasa, Y.; Onji, M. Immune modulator and antiviral potential of dendritic cells pulsed with both hepatitis B surface antigen and core antigen for treating chronic HBV infection. Antivir. Ther. 2010, 15, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Akbar, S.M.F.; Al Mahtab, M.; Aguilar, J.C.; Yoshida, O.; Penton, E.; Gerardo, G.N.; Hiasa, Y. Sustained Antiviral and Liver Protection by a Nasal Therapeutic Vaccine (NASVAC, Containing Both HBsAg and HBcAg) in Patients with Chronic Hepatitis B: 2-Year Follow-Up of Phase III Clinical Trial. Pathogens 2021, 10, 1440. [Google Scholar] [CrossRef]
- Lobaina, Y.; Hardtke, S.; Wedemeyer, H.; Aguilar, J.C.; Schlaphoff, V. In vitro stimulation with HBV therapeutic vaccine candidate Nasvac activates B and T cells from chronic hepatitis B patients and healthy donors. Mol. Immunol. 2015, 63, 320–327. [Google Scholar] [CrossRef]
- Schuurhuis, D.H.; Laban, S.; Toes, R.E.; Ricciardi-Castagnoli, P.; Kleijmeer, M.J.; van der Voort, E.I.H.; Rea, D.; Offring, R.; Geuze, H.J.; Melief, C.J.M.; et al. Immature dendritic cells acquire CD8(+) cytotoxic T lymphocyte priming capacity upon activation by T helper cell-independent or -dependent stimuli. J. Exp. Med. 2000, 192, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Meier, D.; Botter, S.M.; Campanile, C.; Robl, B.; Gräfe, S.; Pellegrini, G.; Born, W.; Fuchs, B. Foscan and foslip based photodynamic therapy in osteosarcoma in vitro and in intratibial mouse models. Int. J. Cancer 2017, 140, 1680–1692. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, C.G.; Camps, M.G.M.; Li, T.M.W.Y.; Chan, A.B.; Ossendorp, F.; Cruz, L.J. Co-delivery of immunomodulators in biodegradable nanoparticles improves therapeutic efficacy of cancer vaccines. Biomaterials 2019, 220, 119417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, J.; Yu, F.S. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 2006, 117, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4⁺ T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926, Erratum in J. Immunother. Cancer 2021, 9, e001926corr1. [Google Scholar] [CrossRef]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef]
- Tanaka, M.; Kataoka, H.; Yano, S.; Sawada, T.; Akashi, H.; Inoue, M.; Suzuki, S.; Inagaki, Y.; Hayashi, N.; Nishie, H.; et al. Immunogenic cell death due to a new photodynamic therapy (PDT) with glycoconjugated chlorin (G-chlorin). Oncotarget 2016, 7, 47242–47251. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Giesselman, B.R.; De Jesús-Andino, F.J.; Foster, T.H. Tumor response to mTHPC-mediated photodynamic therapy exhibits strong correlation with extracellular release of HSP70. Lasers Surg. Med. 2011, 43, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Akbar, S.M.F.; Mahtab, M.A.; Aguilar, J.C.; Uddin, M.H.; Khan, S.I.; Yoshida, O.; Pentón-Arias, E.; Guillen, G.; Hiasa, Y. Repurposing NASVAC, A Hepatitis B Therapeutic Vaccine, for Pre-and Post-exposure Prophylaxis of SARS-CoV-2 Infection. J. Antivir. Antiretrovir. 2021, S20, 004. [Google Scholar]
- Huis In’t Veld, R.V.; Da Silva, C.G.; Jager, M.J.; Cruz, L.J.; Ossendorp, F. Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics 2021, 13, 1470. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Liu, D.; Xu, X.; Ji, J.; Du, Y. Nanomaterials-Based Photodynamic Therapy with Combined Treatment Improves Antitumor Efficacy Through Boosting Immunogenic Cell Death. Int. J. Nanomed. 2021, 16, 4693–4712. [Google Scholar] [CrossRef]
- Pucino, V.; Gardner, D.H.; Fisher, B.A. Rationale for CD40 pathway blockade in autoimmune rheumatic disorders. Lancet Rheumatol. 2020, 2, 292–301. [Google Scholar] [CrossRef]
- Spriggs, M.K.; Armitage, R.J.; Strockbine, L.; Clifford, K.N.; Macduff, B.M.; Sato, T.A.; Maliszewski, C.R.; Fanslow, W.C. Recombinant human CD40 ligand stimulates B cell proliferation and immunoglobulin E secretion. J. Exp. Med. 1992, 176, 1543–1550. [Google Scholar] [CrossRef]
- Kondo, E.; Gryschok, L.; Klein-Gonzalez, N.; Rademacher, S.; Weihrauch, M.R.; Liebig, T.; Shimabukuro-Vornhagen, A.; Kochanek, M.; Draube, A.; Von Bergwelt-Baildon, M.S. CD40-activated B cells can be generated in high number and purity in cancer patients: Analysis of immunogenicity and homing potential. Clin. Exp. Immunol. 2009, 155, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Harada, K.; Horiuchi, S.; Kitamura, D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. eLife 2019, 8, e44245. [Google Scholar] [CrossRef]
- Preise, D.; Oren, R.; Glinert, I.; Kalchenko, V.; Jung, S.; Scherz, A.; Salomon, Y. Systemic antitumor protection by vascular-targeted photodynamic therapy involves cellular and humoral immunity. Cancer Immunol. Immunother. 2009, 58, 71–84. [Google Scholar] [CrossRef]
- Wherry, E.J.; Teichgräber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef]
- Martin, M.D.; Vladimir, P.B. Defining Memory CD8 T Cell. Front. Immunol. 2018, 9, 2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Y.; Gu, Z.; Yu, Z.; Schomann, T.; Sayedipour, S.; Aguilar, J.C.; ten Dijke, P.; Cruz, L.J. Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment. Cancers 2022, 14, 2724. https://doi.org/10.3390/cancers14112724
Hao Y, Gu Z, Yu Z, Schomann T, Sayedipour S, Aguilar JC, ten Dijke P, Cruz LJ. Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment. Cancers. 2022; 14(11):2724. https://doi.org/10.3390/cancers14112724
Chicago/Turabian StyleHao, Yang, Zili Gu, Zhenfeng Yu, Timo Schomann, Sana Sayedipour, Julio C. Aguilar, Peter ten Dijke, and Luis J. Cruz. 2022. "Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment" Cancers 14, no. 11: 2724. https://doi.org/10.3390/cancers14112724
APA StyleHao, Y., Gu, Z., Yu, Z., Schomann, T., Sayedipour, S., Aguilar, J. C., ten Dijke, P., & Cruz, L. J. (2022). Photodynamic Therapy in Combination with the Hepatitis B Core Virus-like Particles (HBc VLPs) to Prime Anticancer Immunity for Colorectal Cancer Treatment. Cancers, 14(11), 2724. https://doi.org/10.3390/cancers14112724