
HydrAd: A Helper-Dependent Adenovirus Targeting Multiple Immune Pathways for Cancer Immunotherapy

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Adenoviral Vectors
2.2. Cell Lines
2.3. Primary Cells
2.4. HDAd Transduction Experiment
2.5. Cytokine/Chemokine ELISA Assay
2.6. Minibody ELISA Assay
2.7. Flow Cytometry
2.8. Co-Culture Experiments
2.9. RNA Extraction and RT-PCR
2.10. MTS Assay
2.11. Statistics and Reproducibility
3. Results
3.1. Switching Ad Serotypes Enables Targeting of a Wide Range of Solid Tumor Cell Lines
3.2. HDAd-Mediated Expression of Immunomodulatory Molecules
3.2.1. Chemokines
3.2.2. Cytokines
3.2.3. Immune Checkpoint Inhibitors
3.2.4. T-Cell Co-Stimulatory Molecules
3.2.5. Engager Molecules
3.3. In-Situ Amplification of HDAd-Encoded Transgenes with Viral-Mediated Oncolysis
3.4. Complex Transgene Constructs
3.4.1. HDTrio
3.4.2. HDTetra
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shaw, A.R.; Suzuki, M. Immunology of Adenoviral Vectors in Cancer Therapy. Mol. Ther. Methods Clin. Dev. 2019, 15, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.; Wilson, J.M.; Batshaw, M.L. Fatal Systemic Inflammatory Response Syndrome in a Ornithine Transcarbamylase Deficient Patient Following Adenoviral Gene Transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus Dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of Virus-Mediated Immunogenic Cancer Cell Death and the Consequences for Oncolytic Virus-Based Immunotherapy of Cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, A.; Nokisalmi, P.; Diaconu, I.; Koski, A.; Cerullo, V.; Liikanen, I.; Tähtinen, S.; Oksanen, M.; Heiskanen, R.; Pesonen, S.; et al. Antiviral and Antitumor T-Cell Immunity in Patients Treated with GM-CSF–Coding Oncolytic Adenovirus. Clin. Cancer Res. 2013, 19, 2734–2744. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-Intrinsic Signaling Pathways: Key Roles in the Regulation of the Immunosuppressive Tumor Microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef]
- Farzad, L.; Cerullo, V.; Yagyu, S.; Bertin, T.; Hemminki, A.; Rooney, C.; Lee, B.; Suzuki, M. Combinatorial Treatment with Oncolytic Adenovirus and Helper-Dependent Adenovirus Augments Adenoviral Cancer Gene Therapy. Mol. Ther. Oncolytics 2014, 1, 14008. [Google Scholar] [CrossRef]
- Tanoue, K.; Shaw, A.R.; Watanabe, N.; Porter, C.; Rana, B.; Gottschalk, S.; Brenner, M.; Suzuki, M. Armed Oncolytic Adenovirus Expressing PD-L1 Mini-Body Enhances Anti-Tumor Effects of Chimeric Antigen Receptor T-Cells in Solid Tumors. Cancer Res. 2017, 77, 2040–2051. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.R.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, C.E.; Shaw, A.R.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.R.; Porter, C.E.; Yip, T.; Mah, W.-C.; McKenna, M.K.; Dysthe, M.; Jung, Y.; Parihar, R.; Brenner, M.K.; Suzuki, M. Oncolytic Adeno-Immunotherapy Modulates the Immune System Enabling CAR T-Cells to Cure Pancreatic Tumors. Commun. Biol. 2021, 4, 368. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Cela, R.; Clarke, C.; Bertin, T.K.; Mouriño, S.; Lee, B. Large-Scale Production of High-Quality Helper-Dependent Adenoviral Vectors Using Adherent Cells in Cell Factories. Hum. Gene Ther. 2010, 21, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Bertin, T.K.; Rogers, G.L.; Cela, R.G.; Zolotukhin, I.; Palmer, D.J.; Ng, P.; Herzog, R.W.; Lee, B. Differential Type I Interferon-Dependent Transgene Silencing of Helper-Dependent Adenoviral vs. Adeno-Associated Viral Vectors In Vivo. Mol. Ther. 2013, 21, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.; Ng, P. Improved System for Helper-Dependent Adenoviral Vector Production. Mol. Ther. 2003, 8, 846–852. [Google Scholar] [CrossRef]
- Guse, K.; Suzuki, M.; Sule, G.; Bertin, T.K.; Tyynismaa, H.; Ahola-Erkkilä, S.; Palmer, D.; Suomalainen, A.; Ng, P.; Cerullo, V.; et al. Capsid-Modified Adenoviral Vectors for Improved Muscle-Directed Gene Therapy. Hum. Gene Ther. 2012, 23, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T Cell Therapy for Breast Cancer: Harnessing the Tumor Milieu to Drive T Cell Activation. J. Immunother. Cancer 2018, 6, 34. [Google Scholar] [CrossRef]
- Gerdemann, U.; Keirnan, J.M.; Katari, U.L.; Yanagisawa, R.; Christin, A.S.; Huye, L.E.; Perna, S.K.; Ennamuri, S.; Gottschalk, S.; Brenner, M.K.; et al. Rapidly Generated Multivirus-Specific Cytotoxic T Lymphocytes for the Prophylaxis and Treatment of Viral Infections. Mol. Ther. 2012, 20, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Vasileiou, S.; Lulla, P.D.; Tzannou, I.; Watanabe, A.; Kuvalekar, M.; Callejas, W.L.; Bilgi, M.; Wang, T.; Wu, M.J.; Kamble, R.; et al. T-Cell Therapy for Lymphoma Using Nonengineered Multiantigen-Targeted T Cells Is Safe and Produces Durable Clinical Effects. J. Clin. Oncol. 2021, 39, 1415–1425. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.C.; Springer, K.; Zhang, Y.B.; Freimuth, P.; Flanagan, J.M. Structural Analysis of the Mechanism of Adenovirus Binding to Its Human Cellular Receptor, CAR. Science 1999, 286, 1579–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Takenobu, H.; Shimozato, O.; Kawamura, K.; Nimura, Y.; Seki, N.; Uzawa, K.; Tanzawa, H.; Shimada, H.; Ochiai, T.; et al. Increased Infectivity of Adenovirus Type 5 Bearing Type 11 or Type 35 Fibers to Human Esophageal and Oral Carcinoma Cells. Oncol. Rep. 2005, 14, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S.; Do, M.-H.; Kwon, S.-Y.; Moon, C.; Kim, K.; Lee, K.; Lee, S.-J.; Hemmi, S.; Joo, Y.-E.; Kim, M.S.; et al. Efficacy of CD46-Targeting Chimeric Ad5/35 Adenoviral Gene Therapy for Colorectal Cancers. Oncotarget 2016, 7, 38210–38223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, M.-H.; To, P.K.; Cho, Y.-S.; Kwon, S.-Y.; Hwang, E.C.; Choi, C.; Cho, S.-H.; Lee, S.-J.; Hemmi, S.; Jung, C. Targeting CD46 Enhances Anti-Tumoral Activity of Adenovirus Type 5 for Bladder Cancer. Int. J. Mol. Sci. 2018, 19, 2694. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Z.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 Is a Receptor for Adenovirus Serotypes 3, 7, 11, and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef]
- Trinh, H.V.; Lesage, G.; Chennamparampil, V.; Vollenweider, B.; Burckhardt, C.J.; Schauer, S.; Havenga, M.; Greber, U.F.; Hemmi, S. Avidity Binding of Human Adenovirus Serotypes 3 and 7 to the Membrane Cofactor CD46 Triggers Infection. J. Virol 2012, 86, 1623–1637. [Google Scholar] [CrossRef] [Green Version]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of Cancer Patients with a Serotype 5/3 Chimeric Oncolytic Adenovirus Expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef]
- Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T.; et al. A Phase I Clinical Trial of Ad5/3-Δ24, a Novel Serotype-Chimeric, Infectivity-Enhanced, Conditionally-Replicative Adenovirus (CRAd), in Patients with Recurrent Ovarian Cancer. Gynecol. Oncol. 2013, 130, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.R.; Suzuki, M. Recent Advances in Oncolytic Adenovirus Therapies for Cancer. Curr. Opin. Virol. 2016, 21, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Tumor-Localized Secretion of Soluble PD1 Enhances Oncolytic Virotherapy. Cancer Res. 2017, 77, 2952–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 2020, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, F.; Bobanga, I.D.; Rauhe, P.; Barkauskas, D.; Teich, N.; Tong, C.; Myers, J.; Huang, A.Y. CCL3 Augments Tumor Rejection and Enhances CD8+ T Cell Infiltration through NK and CD103+ Dendritic Cell Recruitment via IFNγ. Oncoimmunology 2017, 7, e1393598. [Google Scholar] [CrossRef] [Green Version]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective Attraction of Monocytes and T Lymphocytes of the Memory Phenotype by Cytokine RANTES. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, M.B.; Robertson, M.J.; Gordon, M.; Lotze, M.T.; DeCoste, M.; DuBois, J.S.; Ritz, J.; Sandler, A.B.; Edington, H.D.; Garzone, P.D.; et al. Phase I Evaluation of Intravenous Recombinant Human Interleukin 12 in Patients with Advanced Malignancies. Clin. Cancer Res. 1997, 3, 409–417. [Google Scholar] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination Cancer Immunotherapy and New Immunomodulatory Targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Dougan, M.; Rose, N.R.; Lichtman, A.H.; Sharpe, A.H. Adverse Events Following Cancer Immunotherapy: Obstacles and Opportunities. Trends Immunol. 2019, 40, 511–523. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and Coinhibitory Receptors in Anti-Tumor Immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gao, C.; Raymond, M.; Dito, G.; Kabbabe, D.; Shao, X.; Hilt, E.; Sun, Y.; Pak, I.; Gutierrez, M.; et al. An Integrative Approach to Inform Optimal Administration of OX40 Agonist Antibodies in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 6709–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lickliter, J.D.; Hillson, J.L.; Means, G.D.; Sanderson, R.J.; Carley, K.; Tercero, A.; Manjarrez, K.L.; Wiley, J.R.; Peng, S.L. First-in-Human Study of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ALPN-101, a Dual CD28/ICOS Antagonist, in Healthy Adult Subjects. Clin. Transl. Sci. 2021, 14, 1314–1326. [Google Scholar] [CrossRef]
- Nam, K.-O.; Kang, H.; Shin, S.-M.; Cho, K.-H.; Kwon, B.; Kwon, B.S.; Kim, S.-J.; Lee, H.-W. Cross-Linking of 4-1BB Activates TCR-Signaling Pathways in CD8+ T Lymphocytes. J. Immunol. 2005, 174, 1898–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive Immunity Maintains Occult Cancer in an Equilibrium State. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Casucci, M.; di Robilant, B.N.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-Targeted T Cells Mediate Potent Antitumor Effects against Acute Myeloid Leukemia and Multiple Myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for Osteosarcoma: Genetic Modification of T Cells Overcomes Low Levels of Tumor Antigen Expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- De Jaeghere, E.A.; Denys, H.G.; De Wever, O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 2019, 5, 704–723. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer Associated Fibroblasts (CAFs) in Tumor Microenvironment. Front. Biosci 2010, 15, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Santos, L.L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.-H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a Superagonist by Binding to Soluble IL-15R{alpha}. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and Immunogenicity of the ChAdOx1 NCoV-19 Vaccine against SARS-CoV-2: A Preliminary Report of a Phase 1/2, Single-Blind, Randomised Controlled Trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T Cell and Antibody Responses Induced by a Single Dose of ChAdOx1 NCoV-19 (AZD1222) Vaccine in a Phase 1/2 Clinical Trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Boorjian, S.A.; Alemozaffar, M.; Konety, B.R.; Shore, N.D.; Gomella, L.G.; Kamat, A.M.; Bivalacqua, T.J.; Montgomery, J.S.; Lerner, S.P.; Busby, J.E.; et al. Intravesical Nadofaragene Firadenovec Gene Therapy for BCG-Unresponsive Non-Muscle-Invasive Bladder Cancer: A Single-Arm, Open-Label, Repeat-Dose Clinical Trial. Lancet Oncol. 2021, 22, 107–117. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, Y.; Wang, H.; Xu, Z.; Wang, Y.; Li, S.; Liu, J.; Chen, Y.; Luo, H.; Wu, L.; et al. Massive PD-L1 and CD8 Double Positive TILs Characterize an Immunosuppressive Microenvironment with High Mutational Burden in Lung Cancer. J. Immunother. Cancer 2021, 9, e002356. [Google Scholar] [CrossRef]
- Watanabe, N.; McKenna, M.K.; Rosewell Shaw, A.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
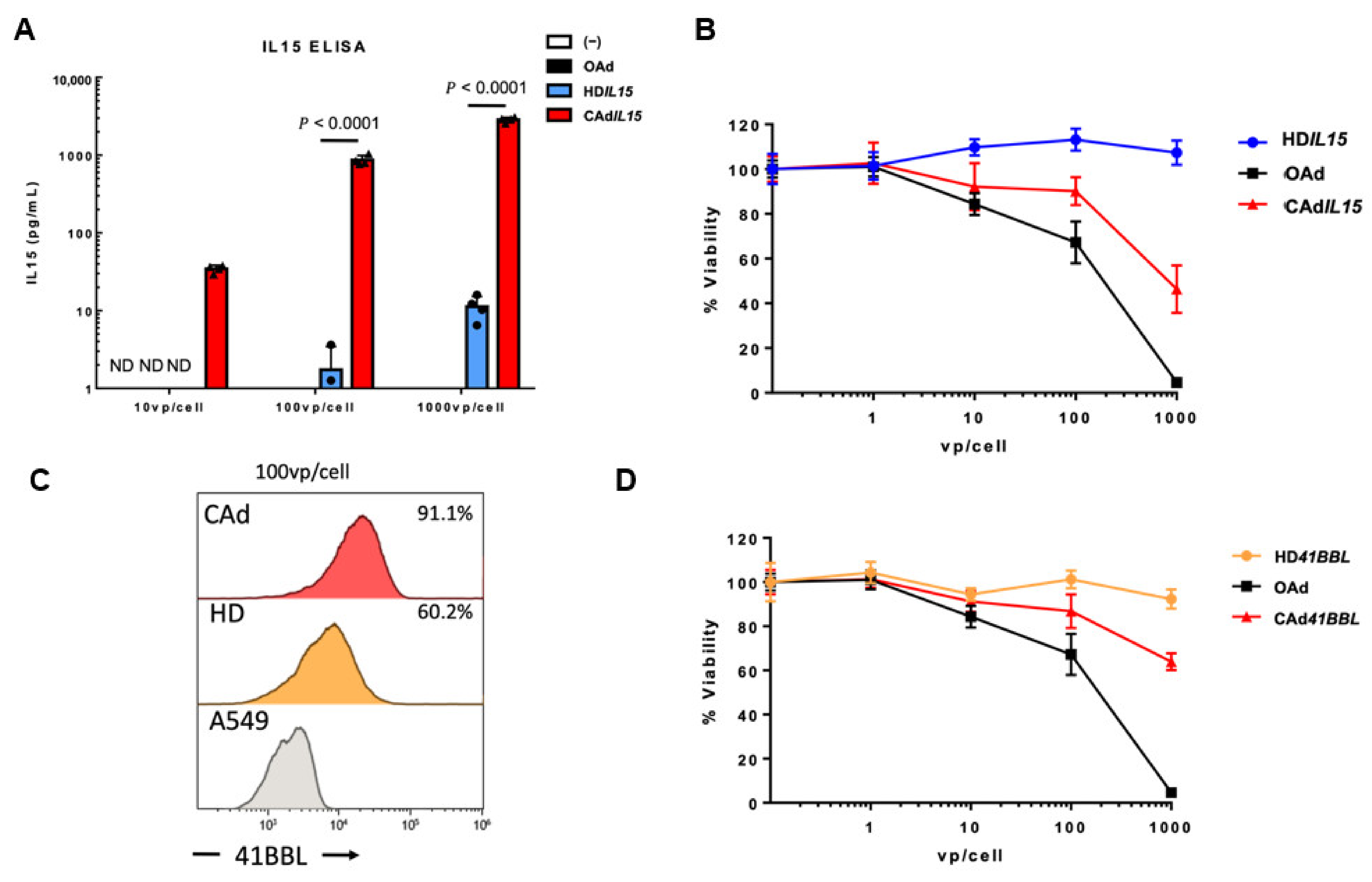{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemokine | Cytokine | Checkpoint Inhibitor | Co-Stimulation | Engager | Suicide Gene |
|---|---|---|---|---|---|
| MIP-1α | IL-2 | αPD-L1 mini-body | 4-1BB ligand | CD19 BiTE | HSTtk |
| RANTES | IL-7 | αPD-1 mini-body | OX40 ligand | CD44v6 BiTE | |
| IL-12p70 | αCTLA-4 mini-body | ICOS ligand | MUC-1 BiTE | ||
| IL-15 | FAP BiTE | ||||
| IL-21 | HER2 BiTE | ||||
| MUC-1 BiKE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosewell Shaw, A.; Porter, C.; Biegert, G.; Jatta, L.; Suzuki, M. HydrAd: A Helper-Dependent Adenovirus Targeting Multiple Immune Pathways for Cancer Immunotherapy. Cancers 2022, 14, 2769. https://doi.org/10.3390/cancers14112769
Rosewell Shaw A, Porter C, Biegert G, Jatta L, Suzuki M. HydrAd: A Helper-Dependent Adenovirus Targeting Multiple Immune Pathways for Cancer Immunotherapy. Cancers. 2022; 14(11):2769. https://doi.org/10.3390/cancers14112769
Chicago/Turabian StyleRosewell Shaw, Amanda, Caroline Porter, Greyson Biegert, Lisa Jatta, and Masataka Suzuki. 2022. "HydrAd: A Helper-Dependent Adenovirus Targeting Multiple Immune Pathways for Cancer Immunotherapy" Cancers 14, no. 11: 2769. https://doi.org/10.3390/cancers14112769
APA StyleRosewell Shaw, A., Porter, C., Biegert, G., Jatta, L., & Suzuki, M. (2022). HydrAd: A Helper-Dependent Adenovirus Targeting Multiple Immune Pathways for Cancer Immunotherapy. Cancers, 14(11), 2769. https://doi.org/10.3390/cancers14112769