
Integrated Multi-Omics Maps of Lower-Grade Gliomas

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
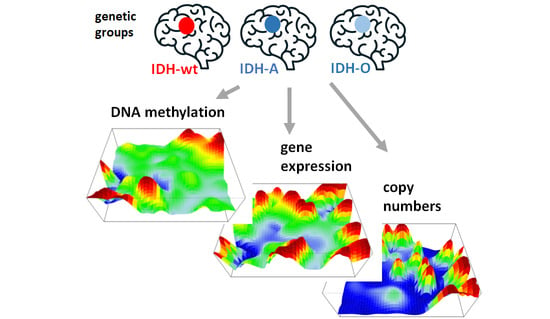
1. Introduction
2. Materials and Methods
2.1. Gene Expression, Methylation, and Copy Number Data of Gliomas
2.2. Preprocessing and Multi-Omics CombiSOM Portrayal
2.3. ScoV (Signed Square Root Covariance) Maps and Mean Portraits
2.4. Spot Module Selection and Functional Analysis
3. Results
3.1. Genetic Stratification of LGG
3.2. Transcriptome, Methylome, and Genome Similarity Patterns of LGG Are Different
3.3. Integrated Portrayal of LGG Reveals Orthogonal Effects of Methylation and CNV
3.4. Cartography of Features, Functions, and of Their Prognostic Impact
3.5. Profiling and Mapping Functional Signatures
3.6. Integrative Portrayal of the LGG Subtype Diversity—Beyond the Genetic Classes
3.7. Reweighting the Modalities—Single Omics Dominated Maps
4. Discussion
4.1. Multi-Omics Cartography of LGG
4.2. LGG Pathogenesis Is Governed by Genetic and Epigenetic Factors along Subtype Specific Paths
4.3. What Modality Is the Best?
4.4. Limitations and Future Applications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| ATAC | assay for transposase-accessible chromatin |
| ATRX | gene encoding ATP-dependent helicase ATRX, X-linked helicase II |
| ChIP-Seq | chromatin immunoprecipitation followed by DNA sequencing |
| Chr | chromosome |
| CIMP | CpG island methylator phenotype in colorectal cancer |
| CpG island | genomics regions with high frequency of cytosine and guanine |
| CTCF^ | gene encoding 11-zinc finger protein or CCCTC-binding factor |
| CVN | copy number variation |
| Dme | DNA methylation |
| DNA | deoxyribonucleic acid |
| E1–E8 | expression subtypes of LGG |
| EMT | epithelial-mesenchymal transition |
| EZH2 | gene encoding Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit, alias KMT6 |
| GBM | Glioblastoma WHO grade IV |
| GCIMP | Glioma CpG-Island hyperMethylation Phenotype |
| GCIMP-O | GCIMP with specific hypermethylation of IDH-O |
| Gex | gene expression |
| GO BP | gene sets related to biological processes; part of Gene Ontology database |
| GPCR | G-protein coupled receptor |
| GSZ | gene set Z score |
| 2HG | 2-hydroxyglutarate, an oncometobolite produced by the mutated IDH enzyme |
| HR | hazard ratio |
| IDH | gene encoding isocitrate dehydrogenase |
| IDH-A | IDH-mutated astrocytoma-like subset of gliomas with chromosome 1p19q intact |
| IDH-mut | gliomas carrying mutation in IDH genes |
| IDH-O | IDH-mutated oligodendroglioma-like subset of gliomas with chromosome 1p19q intact |
| IDH-wt | gliomas with wildtype IDH genes |
| LGG | lower grade diffuse gliomas (WHO grade II and III) |
| M1-M6 | methylation subtypes of LGG |
| MES | mesenchymal subtype of glioblastomas according to Verhaak art al. (2010) [55] |
| MGMT | O-6-Methylguanine-DNA Methyltransferase |
| NL | neuronal-like subset of gliomas according to Verhaak et al. (2010) [55] |
| PRC2 | polycomb repressive complex 2 |
| RTK I/II | GBM methylation class I and II according to Sturm et al. (2012) [41] |
| ScoV | signed square root covariance |
| SOM | self-organizing map |
| SOX2 | gene encoding sex determining region Y (SRY)- box 2 |
| TCGA | The Cancer Genome AtlasTERT telomerase reverse transcriptase |
| TMM | telomere maintenance mechanisms |
| WHO | World Health Organization |
Appendix A. CombiSOM Methods Description
1. Centralization and Harmonization of the Omics Scores
2. CombiSOM Training
3. Signed Square Root Covariance (ScoV) Maps
4. The Gene Set Z score (GSZ) and Ternary GSZ-Diagrams
5. Tumour Similarity Analysis, Supporting and Prognostic Maps
6. Data and Program Availability
Appendix B. Additional Figures and Tables






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot | Brief Characteristics | Up/DN | Top Genes (a) | Gene Sets and p Value of Enrichment (b) |
|---|---|---|---|---|
| A | Verhhak CL/MES_UP | IDH-wt, IDH-A/ IDH-O | INMT, CTHRC1, COL6A3, OAS2, SERPINE, MGP, COL4A2, COL4A1, TNFRSF11, SLC2A10 | WILLSCHER_GBM_Verhaak—CL & MES_up 1 × 10−9 HALLMARK_EPITHELIAL_ MESENCHYMAL_TRANSITION 5 × 10−9, WU_CELL_MIGRATION -08, Phillips MES up vs. Prolif & PN 3 × 10−7 |
| B | GCIMP-meth_UP | IDH-wt/ IDH-A, IDH-O, IDH-A’ | AGAP2, DKK1, TRH, DRC1, MEOX2, MMP9, CHIT1, FMOD, DDIT4L, EMILIN3 | Hopp_Sturm_GBM_ IDH_UP 1 × 10−99, Noushmehr_ GCIMP_hypermeth 2 × 10−22, NOUSHMEHR_GBM_SILENCED_BY_ METHYLATION 5 × 10−22 |
| C | healthy_brain | /IDH- A’ | PDYN, PNOC, SLC13A5, TAC1, TBR1, RYR3, SOSTDC1, PTH2R, PVALB, COL23A | GBM_DN 3 × 10−77, WIRTH_Nervous System 1 × 10−43, Sturm_ RTK II ‘Classic’_UP_RTK I 2 × 10−29, WIRTH_Normal Brain 7 × 10−32 |
| D | Chr. 10− | IDH-A, IDH-A’/ IDH-wt | ARMC3, ITIH2, MCM10, TG, SVIL, IL2RA, MAP3K8, FBXO43, AKR1C2, CCDC3 | Chr 10 1 × 10−99, HOPP_Weak_promoter 4 × 10−13, Reifenberger_GBM_IDH-wt_DN 1 × 10−10 |
| E | Chr. 10− | IDH-A, IDH-O/ IDH-wt | PLEKHS, SFTPD, AFAP1L2, FAM196A, ADRA2A, ATOH7, DUSP5, ADAMTS1, PRLHR, NKX1-2 | Chr 10 1 × 10−99, LASTOWSKA_NEUROBLASTOMA_COPY_ NUMBER_DN 1 × 10−57, Reifenberger_GBM_IDH-wt_DN 1 × 10−32, ROVERSI_GLIOMA_COPY_NUMBER_DN 1 × 10−8 |
| F | Chr.13− | IDH-O/ IDH-A | POSTN, FAM216B, SOX21, GJB2, KL, SGCG, FREM2, PCID2, CUL4A, SKA3 | Chr 13 1 × 10−99 |
| G | Healthy_brain, anti-GCIMP | IDH-O// IDH-wt | NPAS4, EGR4, CHRM1, MARCH4, OPRK1, GPR83, HS3ST3B, SERTM1, SLC32A1, CALB1 | WIRTH_Nervous System 8 × 10−76, GBM_DN 2 × 10−76, WILLSCHER_GBM_Verhaak−PN (mut&wt)_up 2 × 10−60, Sturm_E5_RTK II ‘Classic’_UP 8 × 10−49, Lembcke_TCGA_meth_CIMP.L_UP_CIMP.H_DN 3 × 10−19 |
| H | Chr. 7+ | IDH-wt/ | HOXA5, SLC13A4, WNT2, DLX5, RARRES2, ELN, AZGP1, HOXA7, EGFR, STEAP1 | Chr 7 1 × 10−99, AGUIRRE_PANCREATIC_ CANCER_COPY_NUMBER_UP 8 × 10−10 |
| I | Chr. 19− | IDH-A, IDH-wt/ IDH-O, IDH-A’ | NLRP11, DNAAF3, PRKCG, VSIG10L, FOSB, SYT5, ZNF578, NKG7, FPR3, PPP1R3 | Chr 19 1 × 10−99, KUUSELO_PANCREATIC_ CANCER_19Q13_AMPLIFICATION 5 × 10−35, REACTOME_GENERIC_ TRANSCRIPTION_PATHWAY 4 × 10−34, AGUIRRE_PANCREATIC_ CANCER_COPY_NUMBER_UP 4 × 10−32, ROVERSI_GLIOMA_COPY_NUMBER_UP 8 × 10−24 |
| J | Chr. 1− | IDH-A, IDH-A’, IDH-wt/ IDH-O | WDR63, SPAG17, C1orf194, C1orf58, VAV3, EPHA2, MFAP2, DMRTA2, SLC7A1, RAD54L | Chr 1 1 × 10−99, HOPP_Heterochrom 1 × 10−99, LASTOWSKA_ NEUROBLASTOMA_COPY_NUMBER_DN 1 × 10−99, OKAWA_NEUROBLASTOMA_1P36_31_ DELETION 2 × 10−20, Weller_LGG_A_vs_O_UP 1 × 10−8, Weller_LGG_1p19qDel−vs−intact_DOWN 1 × 10−8 |
| K | GBM_Mesenchymal, Inflammation | IDH-wt, IDH-A; IDH-A’/ IDH-O | CFAP126, COL3A1, C7orf57, METTL7B, CRYBG1, S100A8, CLEC18A, CLEC18C, CLEC18B, CYTL | Sturm_E4_Mesenchymal_RTK I ‘PDGFRA’_DN 1 × 10−99, WILLSCHER_GBM_Verhaak−CL & MES_up 1 × 10−85, Lembcke_TCGA−expr_ CIMP.H_UP 5 × 10−83, CHEN_METABOLIC_ SYNDROM_NETWORK 2 × 10−46, Lembcke_Colonic Inflammation 6 × 10−46, Tirosh_Macrophage specific genes−melanoma 3 × 10−38, immune system process 1 × 10−37 |
References
- Baker, M. Big biology: The ’omes puzzle. Nature 2013, 494, 416–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Loeffler-Wirth, H.; Binder, H. Developmental scRNAseq Trajectories in Gene- and Cell-State Space—The Flatworm Example. Genes 2020, 11, 1214. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Ushijima, T. Compendium of aberrant DNA methylation and histone modifications in cancer. Biochem. Biophys. Res. Commun. 2014, 455, 3–9. [Google Scholar] [CrossRef]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Weisenberger, D.J. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J. Clin. Investig. 2014, 124, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Jeong, J.; Li, L.; Liu, Y.; Nephew, K.P.; Huang, T.H.-M.; Shen, C. An empirical Bayes model for gene expression and methylation profiles in antiestrogen resistant breast cancer. BMC Med. Genom. 2010, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Weichel, B.; Ohm, J.E.; Zhang, K. An integrative analysis of DNA methylation and RNA-Seq data for human heart, kidney and liver. BMC Syst. Biol. 2011, 5, S4. [Google Scholar] [CrossRef] [Green Version]
- Moarii, M.; Boeva, V.; Vert, J.-P.; Reyal, F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genomics 2015, 16, 873. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Louhimo, R.; Hautaniemi, S. CNAmet: An R package for integrating copy number, methylation and expression data. Bioinformatics 2011, 27, 887–888. [Google Scholar] [CrossRef] [Green Version]
- John, C.R.; Watson, D.; Barnes, M.R.; Pitzalis, C.; Lewis, M.J. Spectrum: Fast density-aware spectral clustering for single and multi-omic data. Bioinformatics 2019, 36, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Fujita, N.; Mizuarai, S.; Murakami, K.; Nakai, K. Biomarker discovery by integrated joint non-negative matrix factorization and pathway signature analyses. Sci. Rep. 2018, 8, 9743. [Google Scholar] [CrossRef]
- Yugi, K.; Kubota, H.; Hatano, A.; Kuroda, S. Trans-Omics: How To Reconstruct Biochemical Networks Across Multiple ‘Omic’ Layers. Trends Biotechnol. 2016, 34, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Akhmedov, M.; Arribas, A.; Montemanni, R.; Bertoni, F.; Kwee, I. OmicsNet: Integration of Multi-Omics Data using Path Analysis in Multilayer Networks. bioRxiv 2017, 238766. [Google Scholar]
- Binder, H.; Wirth, H. Analysis of large-scale OMIC data using Self Organizing Maps. In Encyclopedia of Information Science and Technology, 3rd ed.; Khosrow-Pour, M., Ed.; IGI Global: Hershey, PA, USA, 2014; pp. 1642–1654. [Google Scholar]
- Löffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef]
- Hopp, L.; Wirth, H.; Fasold, M.; Binder, H. Portraying the expression landscapes of cancer subtypes: A glioblastoma multiforme and prostate cancer case study. Syst. Biomed. 2013, 1, 99–121. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Reikowski, J.; Hakobyan, S.; Wagner, J.; Binder, H. oposSOM-Browser: An interactive tool to explore omics data landscapes in health science. BMC Bioinform. 2020, 21, 465. [Google Scholar] [CrossRef]
- Weller, M.; Weber, R.G.; Willscher, E.; Riehmer, V.; Hentschel, B.; Kreuz, M.; Felsberg, J.; Beyer, U.; Löffler-Wirth, H.; Kaulich, K.; et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015, 129, 679–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H.; Gietzelt, J.; Hentschel, B.; Westphal, M.; Simon, M.; et al. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Hopp, L.; Willscher, E.; Wirth-Loeffler, H.; Binder, H. Function Shapes Content: DNA-Methylation Marker Genes and their Impact for Molecular Mechanisms of Glioma. J. Cancer Res. Updates 2015, 4, 127–148. [Google Scholar]
- Binder, H.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Jones, D.T.W.; Pfister, S.M.; Kreuz, M.; Gramatzki, D.; Fortenbacher, E.; Hentschel, B.; et al. DNA methylation, transcriptome and genetic copy number signatures of diffuse cerebral WHO grade II/III gliomas resolve cancer heterogeneity and development. Acta Neuropathol. Commun. 2019, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Loeffler-Wirth, H.; Kreuz, M.; Hopp, L.; Arakelyan, A.; Haake, A.; Cogliatti, S.B.; Feller, A.C.; Hansmann, M.; Lenze, D.; Möller, P. A modular transcriptome map of mature B cell lymphomas. Genome Med. 2019, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Hopp, L.; Wirth-Loeffler, H.; Binder, H. Epigenetic heterogeneity of B-cell lymphoma: DNA-methylation, gene expression and chromatin states. Genes 2015, 6, 812–840. [Google Scholar] [CrossRef] [Green Version]
- Hopp, L.; Nersisyan, L.; Löffler-Wirth, H.; Arakelyan, A.; Binder, H. Epigenetic Heterogeneity of B-Cell Lymphoma: Chromatin Modifiers. Genes 2015, 6, 1076. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Jühling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef]
- Binder, H.; Hopp, L.; Lembcke, K.; Wirth, H. Personalized Disease Phenotypes from Massive OMICs Data. In Big Data Analytics in Bioinformatics and Healthcare; Baoying, W., Ruowang, L., William, P., Eds.; IGI Global: Hershey, PA, USA, 2015; pp. 359–378. [Google Scholar]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef]
- Steiner, L.; Hopp, L.; Wirth, H.; Galle, J.; Binder, H.; Prohaska, S.J.; Rohlf, T. A Global Genome Segmentation Method for Exploration of Epigenetic Patterns. PLoS ONE 2012, 7, e46811. [Google Scholar] [CrossRef] [Green Version]
- Fatima, N.; Rueda, L. iSOM-GSN: An integrative approach for transforming multi-omic data into gene similarity networks via self-organizing maps. Bioinformatics 2020, 36, 4248–4254. [Google Scholar] [CrossRef]
- Hopp, L.; Löffler-Wirth, H.; Galle, J.; Binder, H. Combined SOM-portrayal of gene expression and DNA methylation landscapes disentangles modes of epigenetic regulation in glioblastoma. Epigenomics 2018, 10, 745–764. [Google Scholar] [CrossRef]
- Willscher, E.; Hopp, L.; Kreuz, M.; Schmidt, M.; Hakobyan, S.; Arakelyan, A.; Hentschel, B.; Jones, D.T.W.; Pfister, S.M.; Loeffler, M.; et al. High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers 2021, 13, 3198. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Wirth, H.; Löffler, M.; von Bergen, M.; Binder, H. Expression cartography of human tissues using self organizing maps. BMC Bioinformatics 2011, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Wirth, H.; von Bergen, M.; Binder, H. Mining SOM expression portraits: Feature selection and integrating concepts of molecular function. BioData Min. 2012, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Hopp, L.; Lembcke, K.; Binder, H.; Wirth, H. Portraying the Expression Landscapes of B-Cell Lymphoma- Intuitive Detection of Outlier Samples and of Molecular Subtypes. Biology 2013, 2, 1411–1437. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Suvà, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Hopp, L.; Schmidt, M.; Zakharyan, R.; Arakelyan, A.; Binder, H. The Transcriptome and Methylome of the Developing and Aging Brain and Their Relations to Gliomas and Psychological Disorders. Cells 2022, 11, 362. [Google Scholar] [CrossRef]
- Quackenbush, J. Microarrays—Guilt by Association. Science 2003, 302, 240–241. [Google Scholar] [CrossRef]
- Cimino, P.J.; Zager, M.; McFerrin, L.; Wirsching, H.-G.; Bolouri, H.; Hentschel, B.; von Deimling, A.; Jones, D.; Reifenberger, G.; Weller, M.; et al. Multidimensional scaling of diffuse gliomas: Application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol. Commun. 2017, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Buikhuisen, J.Y.; Torang, A.; Medema, J.P. Exploring and modelling colon cancer inter-tumour heterogeneity: Opportunities and challenges. Oncogenesis 2020, 9, 66. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Thalheim, T.; Hopp, L.; Binder, H.; Aust, G.; Galle, J. On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes 2018, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; Gao, L.; Gao, Y.; Hu, Y.; Xu, H.; Huang, M. Evaluation and comparison of multi-omics data integration methods for cancer subtyping. PLoS Comput. Biol. 2021, 17, e1009224. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; van Oudenaarden, A.; Teichmann, S.A. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar] [CrossRef] [Green Version]
- Nersisyan, L.; Loeffler-Wirth, H.; Arakelyan, A.; Binder, H. Gene set- and pathway- centered knowledge discovery assigns transcriptional activation patterns in brain, blood and colon cancer—A bioinformatics perspective. J. Bioinform. Knowl. Min. 2016, 4, 46–70. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binder, H.; Schmidt, M.; Hopp, L.; Davitavyan, S.; Arakelyan, A.; Loeffler-Wirth, H. Integrated Multi-Omics Maps of Lower-Grade Gliomas. Cancers 2022, 14, 2797. https://doi.org/10.3390/cancers14112797
Binder H, Schmidt M, Hopp L, Davitavyan S, Arakelyan A, Loeffler-Wirth H. Integrated Multi-Omics Maps of Lower-Grade Gliomas. Cancers. 2022; 14(11):2797. https://doi.org/10.3390/cancers14112797
Chicago/Turabian StyleBinder, Hans, Maria Schmidt, Lydia Hopp, Suren Davitavyan, Arsen Arakelyan, and Henry Loeffler-Wirth. 2022. "Integrated Multi-Omics Maps of Lower-Grade Gliomas" Cancers 14, no. 11: 2797. https://doi.org/10.3390/cancers14112797
APA StyleBinder, H., Schmidt, M., Hopp, L., Davitavyan, S., Arakelyan, A., & Loeffler-Wirth, H. (2022). Integrated Multi-Omics Maps of Lower-Grade Gliomas. Cancers, 14(11), 2797. https://doi.org/10.3390/cancers14112797