
MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Cell Culture
2.2. LncRNA Subcellular Localization
2.3. Stable MALINC1-Expressing Cells
2.4. Cell Proliferation, Motility, and Migration Assays
2.5. Estradiol Induction Assay
2.6. RNA Isolation and Real-Time Quantitative PCR (RT-qPCR)
2.7. RNA-seq Data Analysis
2.8. In Silico Analysis of MALINC1 Expression in Normal and Breast Cancer Samples
3. Results and Discussion
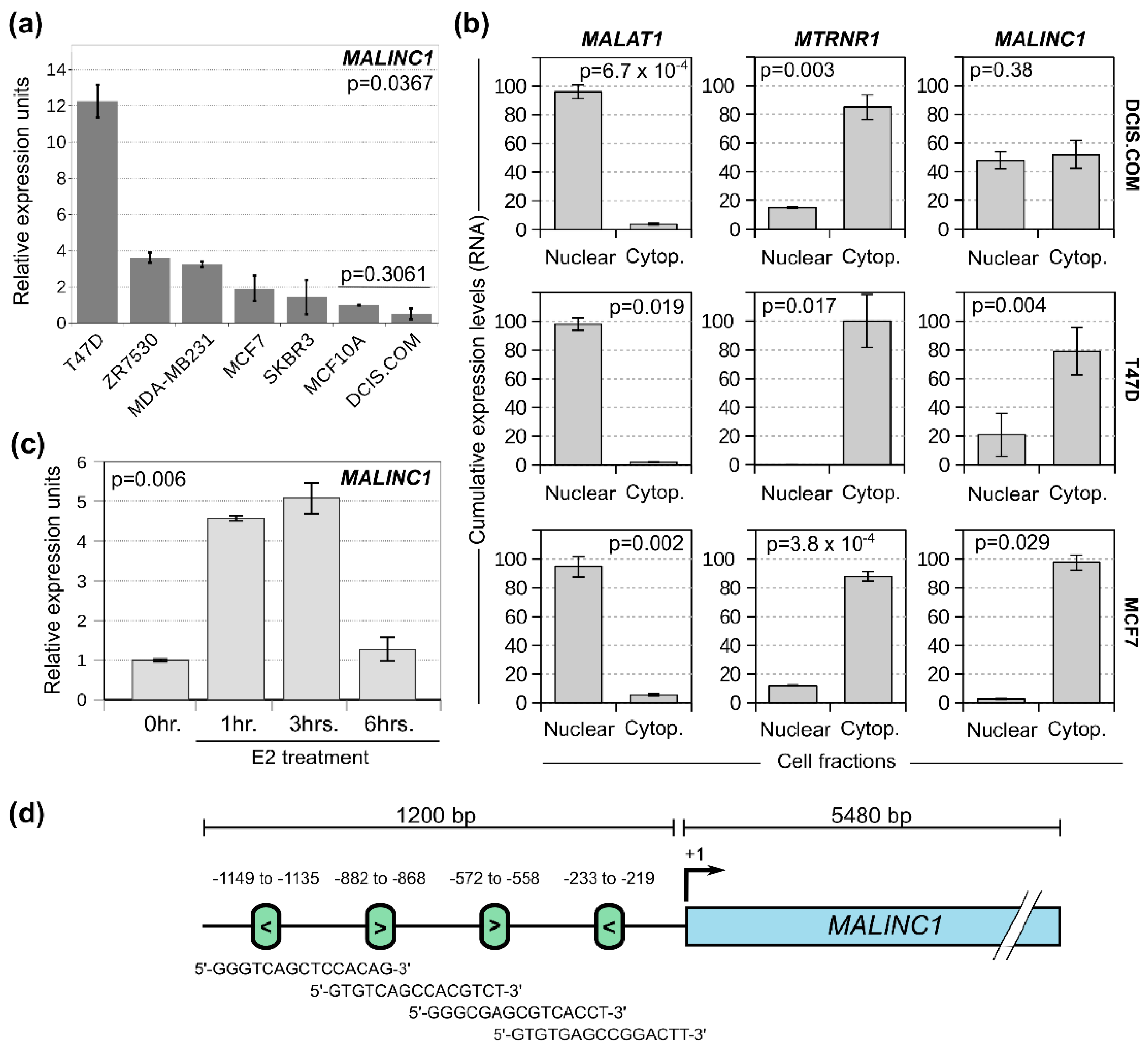
3.1. MALINC1 Is Overexpressed in Luminal Breast Cancer Subtypes and Associated with Poor Prognosis
3.2. MALINC1 Is an E2-ER-Modulated Gene Enriched in the Cytoplasmic Fraction of Luminal Breast Cancer Cells
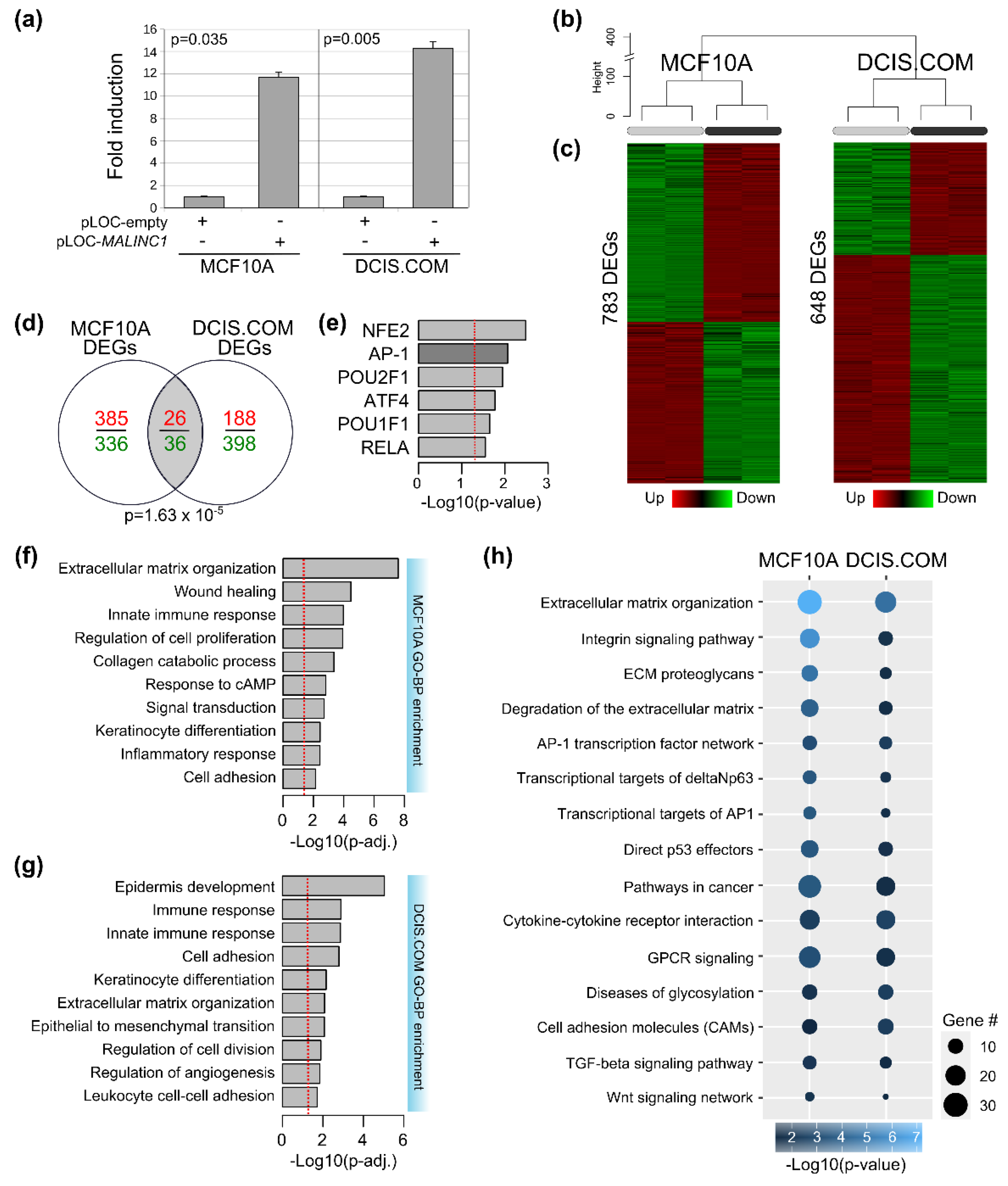
3.3. Transcriptome Analysis of MALINC1-Overexpressing Cells
3.4. MALINC1 Overexpression Promotes In Vitro Migration of Normal Breast Cells
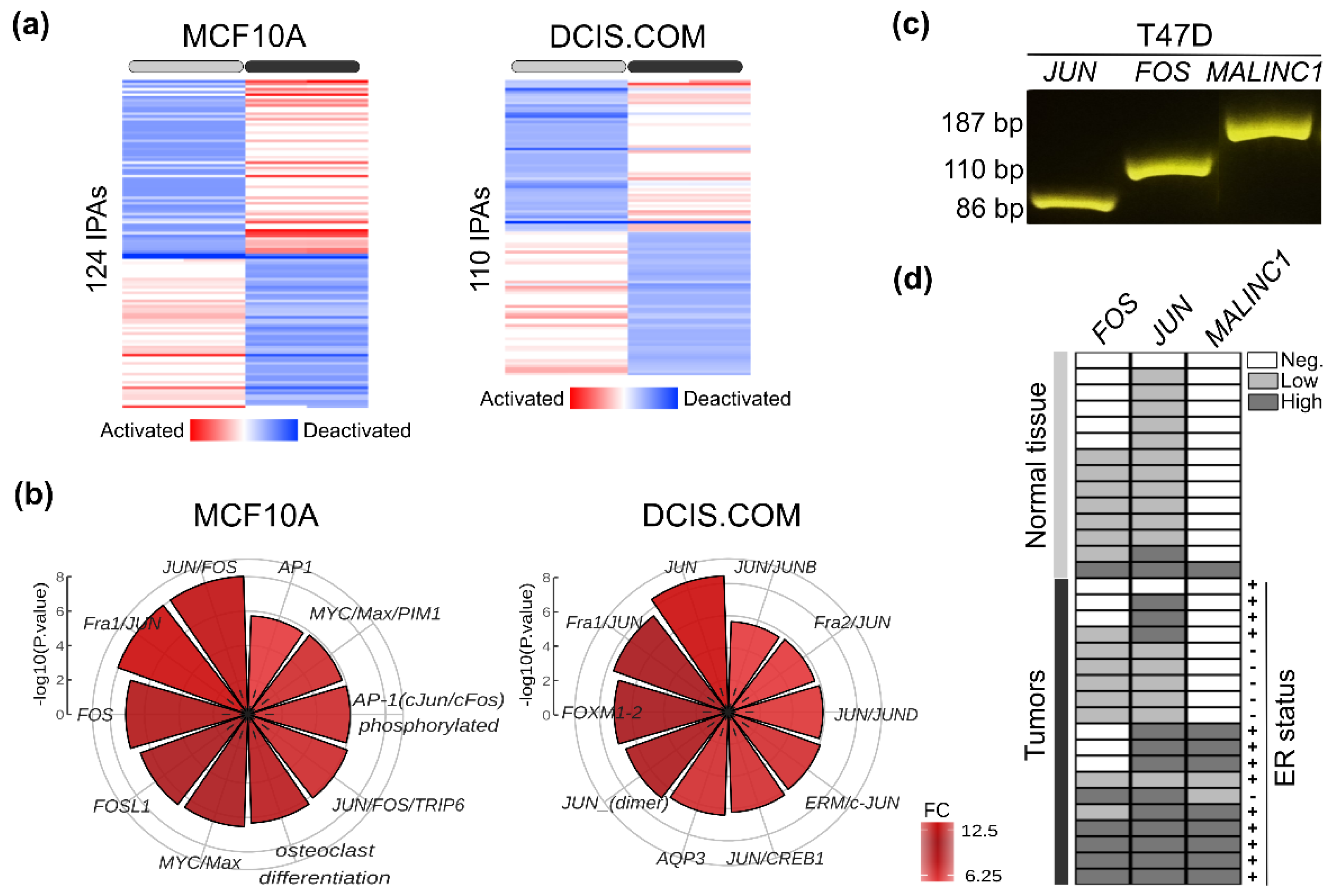
3.5. Conserved MALINC1 Modulated Pathways among Pre-Invasive and Invasive Stages
3.6. Immune Features Associated with MALINC1 Overexpression in Invasive Carcinomas
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wild, C. IARC Publications Website. Available online: http://publications.iarc.fr/586 (accessed on 15 February 2022).
- Invasive Breast Cancer (IDC/ILC). Available online: https://www.cancer.org/cancer/breast-cancer/about/types-of-breast-cancer/invasive-breast-cancer.html (accessed on 23 March 2022).
- Sanders, M.E.; Schuyler, P.A.; Dupont, W.D.; Page, D.L. The natural history of low-grade ductal carcinoma in situ of the breast in women treated by biopsy only revealed over 30 years of long-term follow-up. Cancer 2005, 103, 2481–2484. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Tamimi, R.M.; Baer, H.J.; Connolly, J.L.; Colditz, G.A.; Schnitt, S.J. Outcome of patients with ductal carcinoma in situ untreated after diagnostic biopsy. Cancer 2005, 103, 1778–1784. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Bartel, D.P. LincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Lien, T.; Bergholtz, H.; Fleischer, T.; Djerroudi, L.; Vincent-Salomon, A.; Sørlie, T.; Aittokallio, T. Multi-Omics marker analysis enables early prediction of breast tumor progression. Front. Genet. 2021, 12, 670749. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2018, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ye, C.; Xiong, H.; Shen, Y.; Lu, Y.; Zhou, J.; Wang, L. Dysregulation of long non-coding RNA in breast cancer: An overview of mechanism and clinical implication. Oncotarget 2016, 8, 5508–5522. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Wei, D.; Zhang, Y.; Wang, P.; Zhang, W. Long non-coding rnas as potential diagnostic and prognostic biomarkers in breast cancer: Progress and prospects. Front. Oncol. 2021, 11, 3348. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.C.; Gong, T.; Lu, Y.; Lee, J.; Zhong, Y.; Lacunza, E.; Butti, M.; Takata, Y.; Gaddis, S.; Shen, J.; et al. A molecular portrait of high-grade ductal carcinoma in situ. Cancer Res. 2015, 75, 3980–3990. [Google Scholar] [CrossRef] [Green Version]
- Abba, M.C.; Canzoneri, R.; Gurruchaga, A.; Lee, J.; Tatineni, P.; Kil, H.; Lacunza, E.; Aldaz, C.M. LINC00885 a novel oncogenic long non-coding RNA associated with early stage breast cancer progression. Int. J. Mol. Sci. 2020, 21, 7407. [Google Scholar] [CrossRef]
- Abba, M.C.; Fabre, M.L.; Lee, J.; Tatineni, P.; Kil, H.; Aldaz, C.M. HOTAIR modulated pathways in early-stage breast cancer progression. Front. Oncol. 2021, 11, 4840. [Google Scholar] [CrossRef]
- Bida, O.; Gidoni, M.; Ideses, D.; Efroni, S.; Ginsberg, D. A novel mitosis-associated LncRNA, MA-Linc1, is required for cell cycle progression and sensitizes cancer cells to paclitaxel. Oncotarget 2015, 6, 27880–27890. [Google Scholar] [CrossRef]
- Li, F.-W.; Luo, S.-K. Identification and construction of a predictive immune-related LncRNA signature model for melanoma. Int. J. Gen. Med. 2021, 14, 9227–9235. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Sahoo, D.; Dill, D.L.; Tibshirani, R.; Plevritis, S.K. Extracting binary signals from microarray time-course data. Nucleic Acids Res. 2007, 35, 3705–3712. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Jikuzono, T.; Manabe, E.; Kure, S.; Akasu, H.; Ishikawa, T.; Fujiwara, Y.; Makita, M.; Ishibashi, O. Microarray analysis of ductal carcinoma in situ samples obtained by puncture from surgical resection specimens. BMC Res. Notes 2021, 14, 340. [Google Scholar] [CrossRef]
- DeVaux, R.S.; Ropri, A.S.; Grimm, S.L.; Hall, P.A.; Herrera, E.O.; Chittur, S.V.; Smith, W.P.; Coarfa, C.; Behbod, F.; Herschkowitz, J.I. Long noncoding RNA BHLHE40-AS1 promotes early breast cancer progression through modulating IL-6/STAT3 signaling. J. Cell. Biochem. 2020, 121, 3465–3478. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.H.; Kim, K.M.; McClusky, W.G.; Abdelmohsen, K.; Gorospe, M. Cytoplasmic functions of long noncoding RNAs. WIREs RNA 2018, 9, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.C.; Hu, Y.; Sun, H.; Drake, J.A.; Gaddis, S.; Baggerly, K.; Sahin, A.; Aldaz, C.M. Gene expression signature of estrogen receptor α status in breast cancer. BMC Genom. 2005, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavat, H.; Kurzer, M.S. Estrogen metabolism and breast cancer. Cancer Lett. 2015, 356, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Starek-Świechowicz, B.; Budziszewska, B.; Starek, A. Endogenous Estrogens—Breast cancer and chemoprevention. Pharmacol. Rep. 2021, 73, 1497–1512. [Google Scholar] [CrossRef]
- Murphy, L.C.; Watson, P. Steroid receptors in human breast tumorigenesis and breast cancer progression. Biomed. Pharmacother. 2002, 56, 65–77. [Google Scholar] [CrossRef]
- Clarke, R.B.; Anderson, E.; Howell, A. Steroid receptors in human breast cancer. Trends Endocrinol. Metab. 2004, 15, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 135–170. [Google Scholar]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Medica 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Lesurf, R.; Aure, M.R.; Mørk, H.H.; Vitelli, V.; Lundgren, S.; Børresen-Dale, A.-L.; Kristensen, V.; Wärnberg, F.; Hallett, M.; Sørlie, T.; et al. Molecular features of subtype-specific progression from ductal carcinoma in situ to invasive breast cancer. Cell Rep. 2016, 16, 1166–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, V.N.; Vaske, C.J.; Ursini-Siegel, J.; Van Loo, P.; Nordgard, S.H.; Sachidanandam, R.; Sørlie, T.; Wärnberg, F.; Haakensen, V.D.; Helland, Å.; et al. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 2802–2807. [Google Scholar] [CrossRef] [Green Version]
- Trinh, A.; Gil Del Alcazar, C.R.; Shukla, S.A.; Chin, K.; Chang, Y.H.; Thibault, G.; Eng, J.; Jovanović, B.; Aldaz, C.M.; Park, S.Y.; et al. Genomic alterations during the in situ to invasive ductal breast carcinoma transition shaped by the immune system. Mol. Cancer Res. MCR 2021, 19, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Done, S.J.; Eskandarian, S.; Bull, S.; Redston, M.; Andrulis, I.L. P53 missense mutations in Microdissected High-Grade ductal carcinoma in situ of the breast. JNCI J. Natl. Cancer Inst. 2001, 93, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, C.P.; Mirza, S.; Sharma, G.; Prashad, R.; DattaGupta, S.; Rath, G.; Ralhan, R. Epigenetic alterations of CDH1 and APC Genes: Relationship with activation of Wnt/β-Catenin pathway in invasive ductal carcinoma of breast. Life Sci. 2008, 83, 318–325. [Google Scholar] [CrossRef]
- Varma, K.; Chauhan, A.; Bhargava, M.; Misra, V.; Srivastava, S. Association of Different patterns of expression of beta-catenin and cyclin D1 with pathogenesis of breast carcinoma. Indian J. Pathol. Microbiol. 2020, 63, 13–18. [Google Scholar] [CrossRef]
- Nakamura, R.; Oyama, T.; Inokuchi, M.; Ishikawa, S.; Hirata, M.; Kawashima, H.; Ikeda, H.; Dobashi, Y.; Ooi, A. The relation between Anti-TGBFR1 immunohistochemical reaction and low Ki67, small tumor size and high estrogen receptor expression in invasive breast cancer. Pathol. Int. 2020, 70, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Adhikary, G.; Young, C.A.; Jans, R.; Crish, J.F.; Xu, W.; Rorke, E.A. AP1 transcription factors in epidermal differentiation and skin cancer. J. Skin Cancer 2013, 2013, 537028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 2017, 68, 1067–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375. [Google Scholar] [CrossRef]
- Baan, B.; Pardali, E.; ten Dijke, P.; van Dam, H. In situ proximity ligation detection of C-Jun/AP-1 dimers reveals increased levels of c-Jun/Fra1 complexes in aggressive breast cancer cell lines in vitro and in vivo. Mol. Cell. Proteom. 2010, 9, 1982–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Zhu, B.; Qiu, Z.; Lin, Z. Systematic identification of the key candidate genes in breast cancer stroma. Cell. Mol. Biol. Lett. 2018, 23, 44. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to AP-1. J. Leukoc. Biol. 1998, 63, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription factors as regulators of immune responses in cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 is a key regulator of proinflammatory cytokine tnfα-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016, 291, 5068–5079. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Nichols, J.E.; Valdez, R.; Mendelson, C.R.; Simpson, E.R. Tumor necrosis factor-alpha stimulates aromatase gene expression in human adipose stromal cells through use of an activating protein-1 binding site upstream of promoter 1.4. Mol. Endocrinol. 1996, 10, 1350–1357. [Google Scholar] [CrossRef] [Green Version]
- Mercogliano, M.F.; Bruni, S.; Elizalde, P.V.; Schillaci, R. Tumor necrosis factor α blockade: An opportunity to tackle breast cancer. Front. Oncol. 2020, 10, 584. [Google Scholar] [CrossRef] [Green Version]
- Macián, F.; López-Rodríguez, C.; Rao, A. Partners in Transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8 + T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathas, S. Aberrantly expressed C-Jun and JunB are a hallmark of hodgkin lymphoma cells, stimulate proliferation and synergize with NF-KappaB. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 Activity and EBV infection induce PD-L1 in hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil Del Alcazar, C.R.; Huh, S.J.; Ekram, M.B.; Trinh, A.; Liu, L.L.; Beca, F.; Zi, X.; Kwak, M.; Bergholtz, H.; Su, Y.; et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. 2017, 7, 1098–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The use of immune checkpoint inhibitors in oncology and the occurrence of AKI: Where do we stand? Front. Immunol. 2020, 11, 4271. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisseret, L.; Garaud, S.; de Wind, A.; Van den Eynden, G.; Boisson, A.; Solinas, C.; Gu-Trantien, C.; Naveaux, C.; Lodewyckx, J.-N.; Duvillier, H.; et al. Tumor-Infiltrating lymphocyte composition, organization and PD-1/ PD-L1 expression are linked in breast cancer. OncoImmunology 2016, 6, e1257452. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabre, M.L.; Canzoneri, R.; Gurruchaga, A.; Lee, J.; Tatineni, P.; Kil, H.; Lacunza, E.; Aldaz, C.M.; Abba, M.C. MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression. Cancers 2022, 14, 2819. https://doi.org/10.3390/cancers14122819
Fabre ML, Canzoneri R, Gurruchaga A, Lee J, Tatineni P, Kil H, Lacunza E, Aldaz CM, Abba MC. MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression. Cancers. 2022; 14(12):2819. https://doi.org/10.3390/cancers14122819
Chicago/Turabian StyleFabre, María Laura, Romina Canzoneri, Agustina Gurruchaga, Jaeho Lee, Pradeep Tatineni, Hyunsuk Kil, Ezequiel Lacunza, C. Marcelo Aldaz, and Martín Carlos Abba. 2022. "MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression" Cancers 14, no. 12: 2819. https://doi.org/10.3390/cancers14122819
APA StyleFabre, M. L., Canzoneri, R., Gurruchaga, A., Lee, J., Tatineni, P., Kil, H., Lacunza, E., Aldaz, C. M., & Abba, M. C. (2022). MALINC1 an Immune-Related Long Non-Coding RNA Associated with Early-Stage Breast Cancer Progression. Cancers, 14(12), 2819. https://doi.org/10.3390/cancers14122819