
Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies—A Review

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Paediatric Cancer Incidence
3. Syndromes Predisposing to Haematological Malignancies
3.1. DNA Repair Disorders
3.1.1. Ataxia Teleangiectasia (A-T)
3.1.2. Nijmegen Breakage Syndrome (NBS)
3.1.3. Bloom’s Syndrome (BS)
3.1.4. Constitutional Mismatch Repair Deficiency (CMMRD)
3.1.5. Xeroderma Pigmentosum (XP)
3.2. Bone Marrow Failure
3.2.1. Fanconi Anaemia (FA)
3.2.2. Dyskeratosis Congenita (DC)
3.2.3. Shwachman Diamond Syndrome (SDS)
3.2.4. Diamond Blackfan Anemia (DBA)
3.2.5. GATA2 Deficiency
3.3. Immunodeficiencies with Associated or Syndrome Features
3.3.1. Cartilage–Hair Hypoplasia (CHH)
3.3.2. Wiskott-Aldrich Syndrome (WAS)
3.3.3. SAMD9 and SAMD9L Syndromes
3.4. RASopathies
3.4.1. Noonan Syndrome
3.4.2. Neurofibromatosis Type 1 (NF1)
3.4.3. Casitas B-Lineage Lymphoma (CBL-Syndrome)
3.5. Aneuploidies
4. Non-Syndromic Germline Variants Predisposing to Malignancies
4.1. Germline Predisposition without a Pre-Existing Haematological Disorder or Organ Dysfunction
4.1.1. Familial AML with CEBPA Mutation
4.1.2. Familial MDS/AML with Mutated DDX41
4.1.3. PAX-5-Associated Leukaemia Predisposition
4.1.4. IKZF1 Susceptibility to ALL
4.1.5. DICER1 Syndrome
4.1.6. Li-Fraumeni Syndrome (LFS)
4.2. Germ Line Predisposition with a Pre-Existing Haematological Disorder
4.2.1. ETV6-Related Familial Neutropenia (Thrombocytopenia, Type 5)
4.2.2. Familial Platelet Disorder with Predisposition for AML
4.2.3. ANKRD26-Related Thrombocytopenia
5. The Importance of Identifying Genetic Predispositions to Paediatric Cancers
5.1. Newborn Screening for CPSs
5.2. Identification of Children with Cancers and Probable CPSs Who Would Benefit from Genetic Counselling
5.3. The Role of Novel Genetic Sequencing (NGS)
6. Incorporating Molecular Findings into Clinic
6.1. Multidisciplinary Cooperation
6.2. The Impact of Diagnosis of Germline Variants Predisposing to Malignancies on Clinical Management
6.3. The Role of Targeted Therapy in Paediatric Cancers
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A-T | Ataxia Teleangiectasia |
| ALL | Acute lymphoblastic leukaemia |
| AML | Acute myeloid leukaemia |
| BMF | Bone marrow failure |
| BS | Bloom’s syndrome |
| CBC | Complete blood count |
| CBL-syndrome | Casitas B-lineage lymphoma |
| CHH | Cartilage hair hypoplasia |
| CMMRD | Constitutional Mismatch Repair Immunodeficiency |
| CNS | Central nervous system |
| CPS | Cancer Predisposition Syndrome |
| CT8M | Constitutional trisomy 8 mosaicism |
| DBA | Diamond Blackfan anaemia |
| DC | Dyskeratosis Congenita |
| DLBCL | Diffuse large B-cell lymphoma |
| DNA | Deoxyribonucleic acid |
| DS | Down syndrome |
| ENT | Ear, Nose and Throat |
| FA | Fanconi Anaemia |
| FISH | Fluorescence in-situ hybridization |
| HL | Hodgkin lymphoma |
| HNPCC | Hereditary non-polyposis colorectal carcinoma |
| HSCT | Haematopoietic stem cell transplantation |
| hTP53rc | Heritable TP53-related cancer syndrome |
| IBMFS | Inherited bone marrow failure syndrome |
| ICL | Interstrand crosslink |
| JMML | Juvenile myelomonocytic leukaemia |
| KS | Klinefelter syndrome |
| LFS | Li-Fraumeni syndrome |
| LIMC | Low- and middle-income country |
| MDS | Myelodysplastic syndrome |
| miRNA | Micro-RNA |
| ML-DS | Myeloid leukaemia of Down syndrome |
| MPD | Myeloproliferative disorder |
| MPN | Myeloproliferative neoplasm |
| MPNST | Malignant peripheral nerve sheath tumour |
| MSI | Microsatellite instability |
| NBS | Nijmegen Breakage syndrome |
| NER | Nucleotide excision repair |
| NF1 | Neurofibromatosis type 1 |
| NGS | Next-generation sequencing |
| NHL | Non-Hodgkin lymphoma |
| NK | Natural killer (cell) |
| NS | Noonan syndrome |
| OS | Overall survival |
| P/LP | Pathogenic/Likely pathogenic |
| PDE | Potentially druggable event |
| PID | Primary immunodeficiency disease |
| RNA | Ribonucleic acid |
| SCE | Sister chromatid exchange |
| SDS | Schwachman Diamond syndrome |
| TAM | Transient abnormal myelopoiesis |
| UDP | Uniparental disomy |
| VUS | Variant of unknown significance |
| WAS | Wiskott–Aldrich syndrome |
| WBMRI | Whole-body magnetic resonance imaging |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WHO | World Health Organization |
| WSP | World Standard Population |
| XP | Xeroderma Pigmentosum |
References
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.; Nichols, K.; Plon, S.; Schiffman, J.; Malkin, D. Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin. Cancer Res. 2017, 23, e1–e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargallo, P.; Oltra, S.; Yáñez, Y.; Juan-Ribelles, A.; Calabria, I.; Segura, V.; Lázaro, M.; Balaguer, J.; Tormo, T.; Dolz, S.; et al. Germline Predisposition to Pediatric Cancer, from Next Generation Sequencing to Medical Care. Cancers 2021, 13, 5339. [Google Scholar] [CrossRef]
- Byrjalsen, A.; Hansen, T.; Stoltze, U.; Mehrjouy, M.; Barnkob, N.; Hjalgrim, L.; Mathiasen, R.; Lautrup, C.; Gregersen, P.; Hasle, H.; et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet. 2020, 16, e1009231. [Google Scholar] [CrossRef] [PubMed]
- Mody, R.; Wu, Y.; Lonigro, R.; Cao, X.; Roychowdhury, S.; Vats, P.; Frank, K.; Prensner, J.; Asangani, I.; Palanisamy, N.; et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 2015, 314, 913. [Google Scholar] [CrossRef]
- Sweet-Cordero, E.; Biegel, J. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.; Wu, G.; Edmonson, M.; Gruber, T.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [Green Version]
- Bloom, M.; Maciaszek, J.; Clark, M.; Pui, C.; Nichols, K. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 2019, 13, 55–70. [Google Scholar] [CrossRef]
- Kattner, P.; Strobel, H.; Khoshnevis, N.; Grunert, M.; Bartholomae, S.; Pruss, M.; Fitzel, R.; Halatsch, M.; Schilberg, K.; Siegelin, M.; et al. Compare and contrast: Pediatric cancer versus adult malignancies. Cancer Metastasis Rev. 2019, 38, 673–682. [Google Scholar] [CrossRef]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 contributors. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet. Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Force, L.; Abdollahpour, I.; Advani, S.; Agius, D.; Ahmadian, E.; Alahdab, F.; Alam, T.; Alebel, A.; Alipour, V.; Allen, C.; et al. The global burden of childhood and adolescent cancer in 2017: An analysis of the Global Burden of Disease Study. Lancet Oncol. 2019, 20, 1211–1225. [Google Scholar] [CrossRef] [Green Version]
- Bhakta, N.; Force, L.; Allemani, C.; Atun, R.; Bray, F.; Coleman, M.; Steliarova-Foucher, E.; Frazier, A.; Robison, L.; Rodriguez-Galindo, C.; et al. Childhood cancer burden: A review of global estimates. Lancet Oncol. 2019, 20, e42–e53. [Google Scholar] [CrossRef] [Green Version]
- Seemanova, E.; Varon, R.; Vejvalka, J.; Jarolim, P.; Seeman, P.; Chrzanowska, K.H.; Digweed, M.; Resnick, I.; Kremensky, I.; Saar, K.; et al. The Slavic NBN Founder Mutation: A Role for Reproductive Fitness? PLoS ONE 2016, 11, e0167984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska-Kusnierz, B.; Pastorczak, A.; Fendler, W.; Wakulinska, A.; Dembowska-Baginska, B.; Heropolitanska-Pliszka, E.; Piątosa, B.; Pietrucha, B.; Kałwak, K.; Ussowicz, M.; et al. Hematopoietic Stem Cell Transplantation Positively Affects the Natural History of Cancer in Nijmegen Breakage Syndrome. Clin. Cancer Res. 2020, 27, 575–584. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Godley, L.; Shimamura, A. Genetic predisposition to hematologic malignancies: Management and surveillance. Blood 2017, 130, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Mangaonkar, A.A.; Patnaik, M.M. Hereditary Predisposition to Hematopoietic Neoplasms: When Bloodline Matters for Blood Cancers. Mayo Clin. Proc. 2020, 95, 1482–1498. [Google Scholar] [CrossRef]
- Bomken, S.; van der Werff Ten Bosch, J.; Attarbaschi, A.; Bacon, C.; Borkhardt, A.; Boztug, K.; Fischer, U.; Hauck, F.; Kuiper, R.; Lammens, T.; et al. Current Understanding and Future Research Priorities in Malignancy Associated With Inborn Errors of Immunity and DNA Repair Disorders: The Perspective of an Interdisciplinary Working Group. Front. Immunol. 2018, 9, 2912. [Google Scholar] [CrossRef]
- Szmyd, B.; Mlynarski, W.; Pastorczak, A. Genetic predisposition to lymphomas: Overview of rare syndromes and inherited familial variants. Mutat. Res./Rev. Mutat. Res. 2021, 788, 108386. [Google Scholar] [CrossRef]
- Suarez, F.; Mahlaoui, N.; Canioni, D.; Andriamanga, C.; Dubois d’Enghien, C.; Brousse, N.; Jais, J.; Fischer, A.; Hermine, O.; Stoppa-Lyonnet, D. Incidence, Presentation, and Prognosis of Malignancies in Ataxia-Telangiectasia: A Report From the French National Registry of Primary Immune Deficiencies. J. Clin. Oncol. 2015, 33, 202–208. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.; McGrath-Morrow, S.; Crawford, T.; Lederman, H. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Os, N.; Jansen, A.; van Deuren, M.; Haraldsson, A.; van Driel, N.; Etzioni, A.; van der Flier, M.; Haaxma, C.; Morio, T.; Rawat, A.; et al. Ataxia-telangiectasia: Immunodeficiency and survival. Clin. Immunol. 2017, 178, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Gathmann, B.; Mahlaoui, N.; Gérard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I.; Kindle, G.; Kuijpers, T.; van Beem, R.; Guzman, D.; et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126.e11. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska, K.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deripapa, E.; Balashov, D.; Rodina, Y.; Laberko, A.; Myakova, N.; Davydova, N.; Gordukova, M.; Abramov, D.; Pay, G.; Shelikhova, L.; et al. Prospective Study of a Cohort of Russian Nijmegen Breakage Syndrome Patients Demonstrating Predictive Value of Low Kappa-Deleting Recombination Excision Circle (KREC) Numbers and Beneficial Effect of Hematopoietic Stem Cell Transplantation (HSCT). Front. Immunol. 2017, 8, 807. [Google Scholar] [CrossRef]
- Piatosa, B.; Wolska-Kuśnierz, B.; Tkaczyk, K.; Heropolitanska-Pliszka, E.; Grycuk, U.; Wakulinska, A.; Gregorek, H. T Lymphocytes in Patients With Nijmegen Breakage Syndrome Demonstrate Features of Exhaustion and Senescence in Flow Cytometric Evaluation of Maturation Pathway. Front. Immunol. 2020, 11, 1319. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Heropolitanska-Pliszka, E.; Pietrucha, B.; Sawicka-Powierza, J.; Bernatowska, E.; Wolska-Kusnierz, B.; Pac, M.; Car, H.; Zalewska, A.; Mikoluc, B. Antioxidant Defense, Redox Homeostasis, and Oxidative Damage in Children with Ataxia Telangiectasia and Nijmegen Breakage Syndrome. Front. Immunol. 2019, 10, 2322. [Google Scholar] [CrossRef]
- Walsh, M.; Chang, V.; Kohlmann, W.; Scott, H.; Cunniff, C.; Bourdeaut, F.; Molenaar, J.; Porter, C.; Sandlund, J.; Plon, S.; et al. Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders. Clin. Cancer Res. 2017, 23, e23–e31. [Google Scholar] [CrossRef] [Green Version]
- Cunniff, C.; Bassetti, J.; Ellis, N. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2016, 8, 4–23. [Google Scholar] [CrossRef]
- Ababou, M. Bloom syndrome and the underlying causes of genetic instability. Mol. Genet. Metab. 2021, 133, 35–48. [Google Scholar] [CrossRef]
- Flanagan, M.; Cunniff, C.M. Bloom Syndrome. In GeneReviews®; Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 2006. [Google Scholar]
- Bakry, D.; Aronson, M.; Durno, C.; Rimawi, H.; Farah, R.; Alharbi, Q.; Alharbi, M.; Shamvil, A.; Ben-Shachar, S.; Mistry, M.; et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur. J. Cancer 2014, 50, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Tabori, U.; Hansford, J.; Achatz, M.; Kratz, C.; Plon, S.; Frebourg, T.; Brugières, L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin. Cancer Res. 2017, 23, e32–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef]
- Ripperger, T.; Schlegelberger, B. Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur. J. Med Genet. 2016, 59, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Durno, C.; Sherman, P.; Aronson, M.; Malkin, D.; Hawkins, C.; Bakry, D.; Bouffet, E.; Gallinger, S.; Pollett, A.; Campbell, B.; et al. Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur. J. Cancer 2015, 51, 977–983. [Google Scholar] [CrossRef]
- Guerrini-Rousseau, L.; Varlet, P.; Colas, C.; Andreiuolo, F.; Bourdeaut, F.; Dahan, K.; Devalck, C.; Faure-Conter, C.; Genuardi, M.; Goldberg, Y.; et al. Constitutional mismatch repair deficiency–associated brain tumors: Report from the European C4CMMRD consortium. Neuro Oncol. Adv. 2019, 1, 1–13. [Google Scholar] [CrossRef]
- Martens, M.; Emmert, S.; Boeckmann, L. Xeroderma Pigmentosum: Gene Variants and Splice Variants. Genes 2021, 12, 1173. [Google Scholar] [CrossRef]
- Lehmann, J.; Seebode, C.; Martens, M.; Emmert, S. Xeroderma pigmentosum—Facts and Perspectives. Aktuelle Dermatol. 2018, 44, 232–236. [Google Scholar] [CrossRef]
- Martens, M.C.; Emmert, S.; Boeckmann, L. Sunlight, Vitamin D, and Xeroderma Pigmentosum. Adv. Exp. Med. Biol. 2020, 1268, 319–331. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Lee, M.M.; Andrews, A.D.; Lambert, W.C. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Dermatol. 1994, 130, 1018–1021. [Google Scholar] [CrossRef]
- Seif, A.E. Pediatric leukemia predisposition syndromes: Clues to understanding leukemogenesis. Cancer Genet. 2011, 204, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Rothblum-Oviatt, C.; Ellis, N.; Hickson, I.; Meyer, S.; Crawford, T.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Alter, B. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulliamy, T.; Marrone, A.; Szydlo, R.; Walne, A.; Mason, P.; Dokal, I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004, 36, 447–449. [Google Scholar] [CrossRef] [Green Version]
- Garofola, C.; Nassereddin, A.; Gross, G.P. Dyskeratosis Congenita. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507710/ (accessed on 2 July 2022).
- AlSabbagh, M. Dyskeratosis congenita: A literature review. J. Dtsch. Dermatol. Ges. 2020, 18, 943–967. [Google Scholar] [CrossRef]
- Bezzerri, V.; Cipolli, M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol. Diagn. Ther. 2018, 23, 281–290. [Google Scholar] [CrossRef]
- Nelson, A.; Myers, K. Shwachman-Diamond Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1756/ (accessed on 2 April 2022).
- Myers, K.C.; Furutani, E.; Weller, E.; Siegele, B.; Galvin, A.; Arsenault, V.; Alter, B.P.; Boulad, F.; Bueso-Ramos, C.; Burroughs, L.; et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: A multicentre, retrospective, cohort study. Lancet Haematol. 2020, 7, e238–e246. [Google Scholar] [CrossRef]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, L.; Leblanc, T.; Mohandas, N. Diamond-Blackfan anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef]
- Bartels, M.; Bierings, M. How I manage children with Diamond-Blackfan anaemia. Br. J. Haematol. 2018, 184, 123–133. [Google Scholar] [CrossRef]
- Vlachos, A.; Rosenberg, P.; Atsidaftos, E.; Alter, B.; Lipton, J. Incidence of neoplasia in Diamond Blackfan anemia: A report from the Diamond Blackfan Anemia Registry. Blood 2012, 119, 3815–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babushok, D.; Bessler, M.; Olson, T. Genetic predisposition to myelodysplastic syndrome and acute myeloid leukemia in children and young adults. Leuk. Lymphoma 2015, 57, 520–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klco, J.; Mullighan, C. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat. Rev. Cancer 2020, 21, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Druley, T.; Erez, A.; Kuiper, R.; Onel, K.; Schiffman, J.; Wolfe Schneider, K.; Scollon, S.; Scott, H.; Strong, L.; et al. Recommendations for Surveillance for Children with Leukemia-Predisposing Conditions. Clin. Cancer Res. 2017, 23, e14–e22. [Google Scholar] [CrossRef] [Green Version]
- Vakkilainen, S.; Taskinen, M.; Mäkitie, O. Immunodeficiency in cartilage-hair hypoplasia: Pathogenesis, clinical course and management. Scand. J. Immunol. 2020, 92, e12913. [Google Scholar] [CrossRef]
- Makitie, O. Cartilage-hair hypoplasia in Finland: Epidemiological and genetic aspects of 107 patients. J. Med Genet. 1992, 29, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Taskinen, M.; Ranki, A.; Pukkala, E.; Jeskanen, L.; Kaitila, I.; Mäkitie, O. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am. J. Med Genet. Part A 2008, 146A, 2370–2375. [Google Scholar] [CrossRef]
- Candotti, F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 2017, 38, 13–27. [Google Scholar] [CrossRef]
- Leechawengwongs, E.; Shearer, W. Lymphoma complicating primary immunodeficiency syndromes. Curr. Opin. Hematol. 2012, 19, 305–312. [Google Scholar] [CrossRef]
- Duan, L.; Grunebaum, E. Hematological Malignancies Associated With Primary Immunodeficiency Disorders. Clin. Immunol. 2018, 194, 46–59. [Google Scholar] [CrossRef]
- Sahoo, S.; Kozyra, E.; Wlodarski, M. Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract. Res. Clin. Haematol. 2020, 33, 101197. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Below, J.; Shimamura, A.; Keel, S.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A. Noonan Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1124/ (accessed on 2 April 2022).
- Villani, A.; Greer, M.; Kalish, J.; Nakagawara, A.; Nathanson, K.; Pajtler, K.; Pfister, S.; Walsh, M.; Wasserman, J.; Zelley, K.; et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin. Cancer Res. 2017, 23, e83–e90. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.; Freedenberg, D.; Schorry, E.; Ullrich, N.; Viskochil, D.; Korf, B.; Chen, E.; Trotter, T.; Berry, S.; Burke, L.; et al. Health Supervision for Children With Neurofibromatosis Type. Pediatrics 2019, 143, e20190660. [Google Scholar] [CrossRef] [Green Version]
- Sur, M.; Armat, I.; Sur, G.; Pop, D.; Samasca, G.; Lupan, I.; Timis, T.; Florian, I.; Sur, D. Neurofibromatosis in Children: Actually and Perspectives. Children 2022, 9, 40. [Google Scholar] [CrossRef]
- Pinti, E.; Nemeth, K.; Staub, K.; Lengyel, A.; Fekete, G.; Haltrich, I. Diagnostic difficulties and possibilities of NF1-like syndromes in childhood. BMC Pediatr. 2021, 21, 331. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef]
- Niemeyer, C. JMML genomics and decisions. Hematology 2018, 2018, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.; Salvador, H.; Chang, V.; Erez, A.; Voss, S.; Schneider, K.; Scott, H.; Plon, S.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis. Clin. Cancer Res. 2017, 23, e46–e53. [Google Scholar] [CrossRef] [Green Version]
- Bülow, L.; Lissewski, C.; Bressel, R.; Rauch, A.; Stark, Z.; Zenker, M.; Bartsch, O. Hydrops, fetal pleural effusions and chylothorax in three patients with CBL mutations. Am. J. Med Genet. Part A 2015, 167A, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Naramura, M.; Nadeau, S.; Mohapatra, B.; Ahmad, G.; Mukhopadhyay, C.; Sattler, M.; Raja, S.M.; Natarajan, A.; Band, V.; Band, H. Mutant Cbl proteins as oncogenic drivers in myeloproliferative disorders. Oncotarget 2011, 2, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, L.; Galán-Gómez, V.; Corral-Sánchez, M.; Pérez-Martínez, A.; Riesco, S.; Isidoro-García, M.; Escudero, A. Juvenile myelomonocytic leukemia in CBL syndrome associated with germline splice-site mutations: Two case reports and a literature review. Clin. Case Rep. 2021, 9, e04260. [Google Scholar] [CrossRef]
- Roberts, I.; Izraeli, S. Haematopoietic development and leukaemia in Down syndrome. Br. J. Haematol. 2014, 167, 587–599. [Google Scholar] [CrossRef]
- Zwaan, C.; Reinhardt, D.; Hitzler, J.; Vyas, P. Acute Leukemias in Children with Down Syndrome. Pediatr. Clin. N. Am. 2008, 55, 53–70. [Google Scholar] [CrossRef]
- Davidsson, J.; Veerla, S.; Johansson, B. Constitutional trisomy 8 mosaicism as a model for epigenetic studies of aneuploidy. Epigenet. Chromatin 2013, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Vo, D.; Mwangi, L.; Rehman, S.; Peiris, A. Oncologic manifestations of Klinefelter syndrome. Hormones 2020, 19, 497–504. [Google Scholar] [CrossRef]
- Ji, J.; Zöller, B.; Sundquist, J.; Sundquist, K. Risk of solid tumors and hematological malignancy in persons with Turner and Klinefelter syndromes: A national cohort study. Int. J. Cancer 2016, 139, 754–758. [Google Scholar] [CrossRef]
- Rau, R.; Carroll, A.; Heerema, N.; Arland, L.; Carroll, W.; Winick, N.; Raetz, E.; Loh, M.; Yang, W.; Relling, M.; et al. Klinefelter syndrome and 47,XYY syndrome in children with B cell acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 179, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Seipel, K.; Pemov, A.; Dewan, R.; Brown, C.; Ravichandran, S.; Luke, B.; Malasky, M.; Suman, S.; Yeager, M.; et al. Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica 2015, 101, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawana, K.; Rio-Machin, A.; Preudhomme, C.; Fitzgibbon, J. Familial CEBPA-mutated acute myeloid leukemia. Semin. Hematol. 2017, 54, 87–93. [Google Scholar] [CrossRef]
- Pabst, T.; Eyholzer, M.; Haefliger, S.; Schardt, J.; Mueller, B.U. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5088–5093. [Google Scholar] [CrossRef]
- Feurstein, S.; Drazer, M.; Godley, L. Genetic predisposition to leukemia and other hematologic malignancies. Semin. Oncol. 2016, 43, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, M.; Brown, A.; Weinel, L.; Phung, C.; Rafidi, G.; Lee, M.; Schreiber, A.; Feng, J.; Babic, M.; Chong, C.; et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood 2016, 127, 1017–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polprasert, C.; Schulze, I.; Sekeres, M.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.; Gu, X.; Phillips, J.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Churchman, M.; Qian, M.; te Kronnie, G.; Zhang, R.; Yang, W.; Zhang, H.; Lana, T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 2018, 33, 937–948.e8. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Bahubeshi, A.; Hamel, N.; Pasini, B.; Asioli, S.; Baynam, G.; Choong, C.S.; Charles, A.; Frieder, R.P.; Dishop, M.K.; et al. Extending the phenotypes associated with DICER1 mutations. Hum. Mutat. 2011, 32, 1381–1384. [Google Scholar] [CrossRef]
- Kuhlen, M.; Hönscheid, A.; Schemme, J.; Merz, H.; Mauz-Körholz, C.; Borkhardt, A.; Troeger, A. Hodgkin lymphoma as a novel presentation of familial DICER1 syndrome. Eur. J. Pediatr. 2015, 175, 593–597. [Google Scholar] [CrossRef]
- Schultz, K.; Rednam, S.P.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.D.; Diller, L.R.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and Their Associated Tumour Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, e76–e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farouk Sait, S.; Walsh, M.; Karajannis, M. Genetic syndromes predisposing to pediatric brain tumors. Neuro-Oncol. Pract. 2021, 8, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Steinke-Lange, V.; Spier, I.; Aretz, S.; Schröck, E.; Holinski-Feder, E. Overview of the Clinical Features of Li-Fraumeni Syndrome and the Current European ERN GENTURIS Guideline. Geburtshilfe Frauenheilkd. 2021, 82, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G.; European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. EJHG 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.; Freycon, C.; Maxwell, K.; Nichols, K.; Schiffman, J.; Evans, D.; Achatz, M.; Savage, S.; Weitzel, J.; Garber, J.; et al. Analysis of the Li-Fraumeni Spectrum Based on an International Germline TP53 Variant Data Set. JAMA Oncol. 2021, 7, 1800. [Google Scholar] [CrossRef]
- Ripperger, T.; Bielack, S.; Borkhardt, A.; Brecht, I.; Burkhardt, B.; Calaminus, G.; Debatin, K.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med Genet. Part A 2017, 173, 1017–1037. [Google Scholar] [CrossRef]
- Feurstein, S.; Godley, L. Germline ETV6 mutations and predisposition to hematological malignancies. Int. J. Hematol. 2017, 106, 189–195. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.; Wedge, D.; Tubio, J.; Alexandrov, L.; Van Loo, P.; Cooke, S.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Bluteau, D.; Balduini, A.; Balayn, N.; Currao, M.; Nurden, P.; Deswarte, C.; Leverger, G.; Noris, P.; Perrotta, S.; Solary, E.; et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J. Clin. Investig. 2014, 124, 580–591. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, G.; Christensen, K.; Sullivan, H.; Stout, N.; Diller, L.; Yeh, J.; Wu, A. Estimated Cost-effectiveness of Genetic Testing in Siblings of Newborns With Cancer Susceptibility Gene Variants. JAMA Netw. Open 2021, 4, e2129742. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.M.; Stout, N.K.; Chaudhry, A.; Christensen, K.D.; Gooch, M.; McMahon, P.M.; O’Brien, G.; Rehman, N.; Blout Zawatsky, C.L.; Green, R.C.; et al. Universal newborn genetic screening for pediatric cancer predisposition syndromes: Model-based insights. Genet. Med. Off. J. Am. Coll. Med Genet. 2021, 23, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Offit, K.; Tkachuk, K.; Stadler, Z.; Walsh, M.; Diaz-Zabala, H.; Levin, J.; Steinsnyder, Z.; Ravichandran, V.; Sharaf, R.; Frey, M.; et al. Cascading After Peridiagnostic Cancer Genetic Testing: An Alternative to Population-Based Screening. J. Clin. Oncol. 2020, 38, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Wolfe Schneider, K.; Jasperson, K. Unique Genetic Counseling Considerations in the Pediatric Oncology Setting. Curr. Genet. Med. Rep. 2015, 3, 65–73. [Google Scholar] [CrossRef]
- NSGC. Policy, Research and Publications. Professional Status Survey. Available online: https://www.nsgc.org/Policy-Research-and-Publications/Professional-Status-Survey (accessed on 2 April 2022).
- Jongmans, M.C.; Loeffen, J.L.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Druker, H.; Zelley, K.; McGee, R.; Scollon, S.; Kohlmann, W.; Schneider, K.; Wolfe Schneider, K. Genetic Counselor Recommendations for Cancer Predisposition Evaluation and Surveillance in the Pediatric Oncology Patient. Clin. Cancer Res. 2017, 23, e91–e97. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.; Gutierrez, S.; Petersen, A.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children with Solid Tumors. JAMA Oncol. 2016, 2, 616. [Google Scholar] [CrossRef]
- Cullinan, N.; Schiller, I.; Di Giuseppe, G.; Mamun, M.; Reichman, L.; Cacciotti, C.; Wheaton, L.; Caswell, K.; Di Monte, B.; Gibson, P.; et al. Utility of a Cancer Predisposition Screening Tool for Predicting Subsequent Malignant Neoplasms in Childhood Cancer Survivors. J. Clin. Oncol. 2021, 39, 3207–3216. [Google Scholar] [CrossRef]
- Goudie, C.; Coltin, H.; Witkowski, L.; Mourad, S.; Malkin, D.; Foulkes, W.D. The McGill Interactive Pediatric OncoGenetic Guidelines: An approach to identifying pediatric oncology patients most likely to benefit from a genetic evaluation. Pediatr. Blood Cancer 2017, 64, e26441. [Google Scholar] [CrossRef]
- Goudie, C.; Witkowski, L.; Cullinan, N.; Reichman, L.; Schiller, I.; Tachdjian, M.; Armstrong, L.; Blood, K.A.; Brossard, J.; Brunga, L.; et al. Performance of the McGill Interactive Pediatric OncoGenetic Guidelines for Identifying Cancer Predisposition Syndromes. JAMA Oncol. 2021, 7, 1806–1814. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, Y.; He, H.; Shen, J. Clinical Interpretation of Sequence Variants. Curr. Protoc. Hum. Genet. 2020, 106, e98. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Martin, N.; Magnus, D. Privacy and ethical challenges in next-generation sequencing. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- McCullough, L.; Slashinski, M.; McGuire, A.; Street, R.; Eng, C.; Gibbs, R.; Parsons, D.; Plon, S. Is Whole-Exome Sequencing an Ethically Disruptive Technology? Perspectives of Pediatric Oncologists and Parents of Pediatric Patients with Solid Tumors. Pediatr. Blood Cancer 2015, 63, 511–515. [Google Scholar] [CrossRef]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discov. 2021, 11, 3008–3027. [Google Scholar] [CrossRef]
- Surrey, L.; MacFarland, S.; Chang, F.; Cao, K.; Rathi, K.; Akgumus, G.; Gallo, D.; Lin, F.; Gleason, A.; Raman, P.; et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Langenberg, K.; Looze, E.J.; Molenaar, J.J. The Landscape of Pediatric Precision Oncology: Program Design, Actionable Alterations, and Clinical Trial Development. Cancers 2021, 13, 4324. [Google Scholar] [CrossRef]
- Marks, L.; Oberg, J.; Pendrick, D.; Sireci, A.; Glasser, C.; Coval, C.; Zylber, R.; Chung, W.; Pang, J.; Turk, A.; et al. Precision Medicine in Children and Young Adults with Hematologic Malignancies and Blood Disorders: The Columbia University Experience. Front. Pediatr. 2017, 5, 265. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Van den Bruel, A.; Bankhead, C.; Mitchell, C.; Phillips, B.; Thompson, M. Clinical presentation of childhood leukaemia: A systematic review and meta-analysis. Arch. Dis. Child. 2016, 101, 894–901. [Google Scholar] [CrossRef]
- Cortelazzo, S.; Ferreri, A.; Hoelzer, D.; Ponzoni, M. Lymphoblastic lymphoma. Crit. Rev. Oncol. Hematol. 2017, 113, 304–317. [Google Scholar] [CrossRef]
- Kratz, C.; Achatz, M.; Brugières, L.; Frebourg, T.; Garber, J.; Greer, M.; Hansford, J.; Janeway, K.; Kohlmann, W.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, A.; Shore, A.; Wasserman, J.; Stephens, D.; Kim, R.; Druker, H.; Gallinger, B.; Naumer, A.; Kohlmann, W.; Novokmet, A.; et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016, 17, 1295–1305. [Google Scholar] [CrossRef]
- Tak, C.; Biltaji, E.; Kohlmann, W.; Maese, L.; Hainaut, P.; Villani, A.; Malkin, D.; Sherwin, C.; Brixner, D.; Schiffman, J. Cost-effectiveness of early cancer surveillance for patients with Li–Fraumeni syndrome. Pediatr. Blood Cancer 2019, 66, e27629. [Google Scholar] [CrossRef] [PubMed]




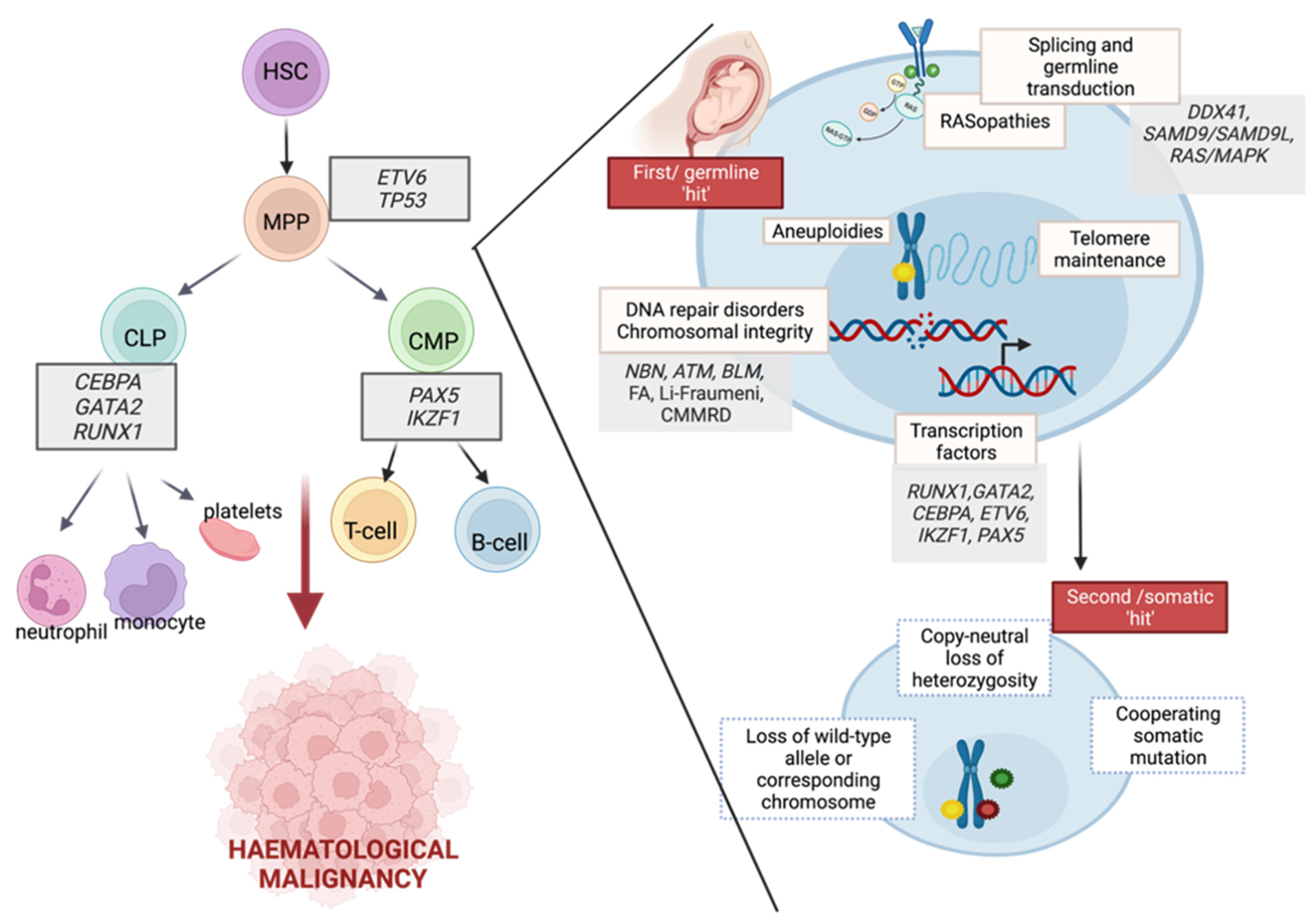{kind=link}
{kind=link}
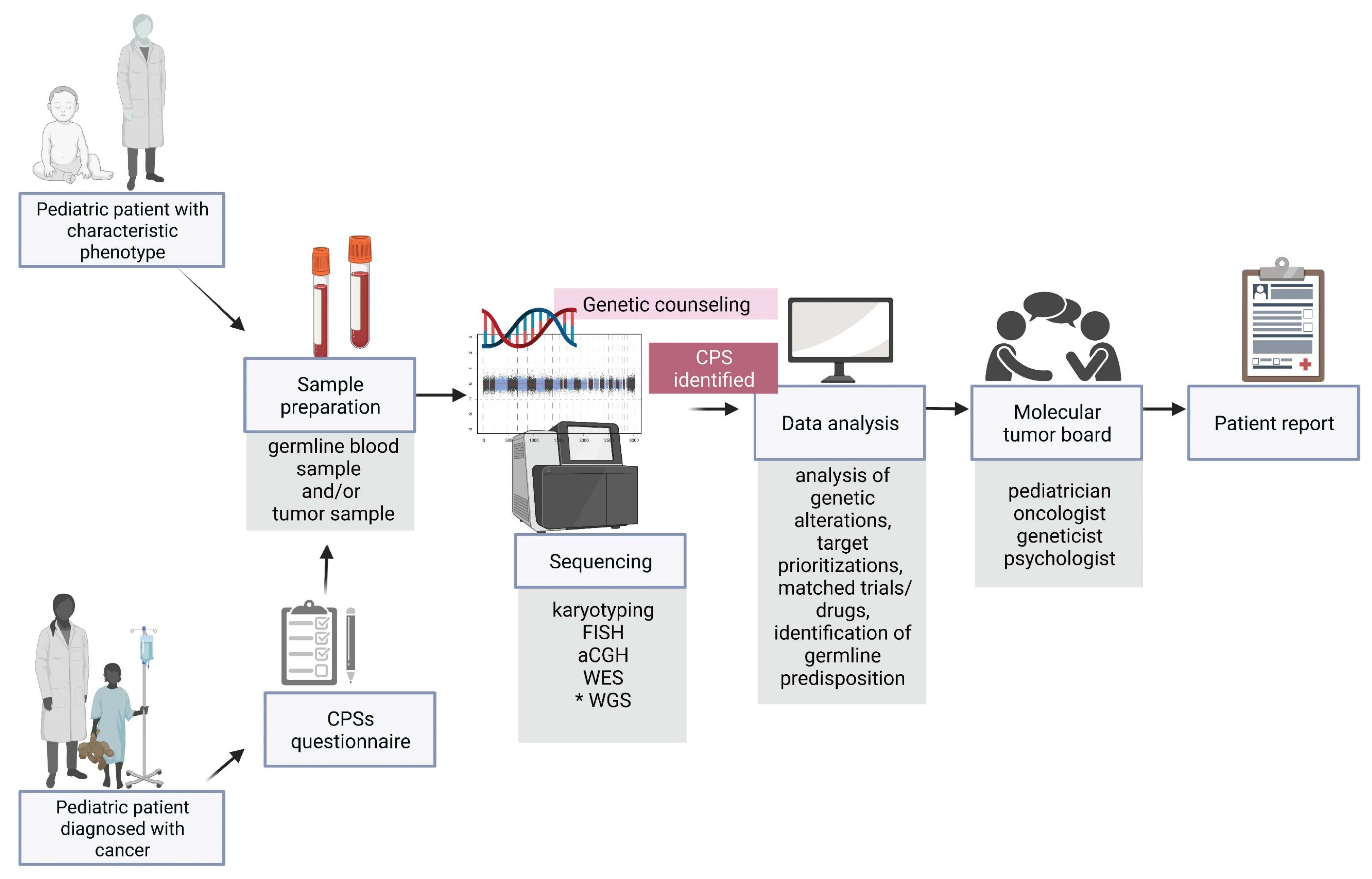{kind=link}
| Syndrome | Patient Care | References |
|---|---|---|
| Li Fraumeni syndrome | Children from birth to age 18: Abdominal and pelvis US every 3–4 m Annually brain MRI (first MRI with contrast) WBMRI annually | [126] |
| Neurofibromatosis 1 | Since birth to age 8: ophthalmology assessment every 6 m to age 1 y Since age 8–20: ophthalmology assessment every 1–2 y At age of 16–20: consider WBMRI | [75] |
| Constitutional mismatch repair deficiency | Since age 6 WBMRI annually From diagnosis of brain tumours brain MRI every 6 m Since age 4 to 6 upper gastrointestinal endoscopy, visual capsule endoscopy, ileocolonoscopy annually | [33] |
| Bloom syndrome | At age 15 colonoscopy every 2 y, faecal occult blood every 6 m At age 20–25 breast MRI/US every 2 y | [28] |
| Xeroderma pigmentosum | Every 3 m skin examination Every 6–12 m exam for ocular and ear, nose, and throat neoplasms | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipiuk, A.; Kozakiewicz, A.; Kośmider, K.; Lejman, M.; Zawitkowska, J. Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies—A Review. Cancers 2022, 14, 3569. https://doi.org/10.3390/cancers14153569
Filipiuk A, Kozakiewicz A, Kośmider K, Lejman M, Zawitkowska J. Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies—A Review. Cancers. 2022; 14(15):3569. https://doi.org/10.3390/cancers14153569
Chicago/Turabian StyleFilipiuk, Aleksandra, Agata Kozakiewicz, Kamil Kośmider, Monika Lejman, and Joanna Zawitkowska. 2022. "Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies—A Review" Cancers 14, no. 15: 3569. https://doi.org/10.3390/cancers14153569
APA StyleFilipiuk, A., Kozakiewicz, A., Kośmider, K., Lejman, M., & Zawitkowska, J. (2022). Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies—A Review. Cancers, 14(15), 3569. https://doi.org/10.3390/cancers14153569