
Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Ethics Statement
2.2. Study Population
2.3. Clinical and Pathological Data
2.4. Mutation Detection
2.5. Statistical Analysis
3. Results
3.1. Clinical Phenotype of Germline Mutation Carriers
3.1.1. ATM Mutation Carriers
3.1.2. CHEK2 Mutation Carriers
3.1.3. HOXB13G84E Mutation Carriers
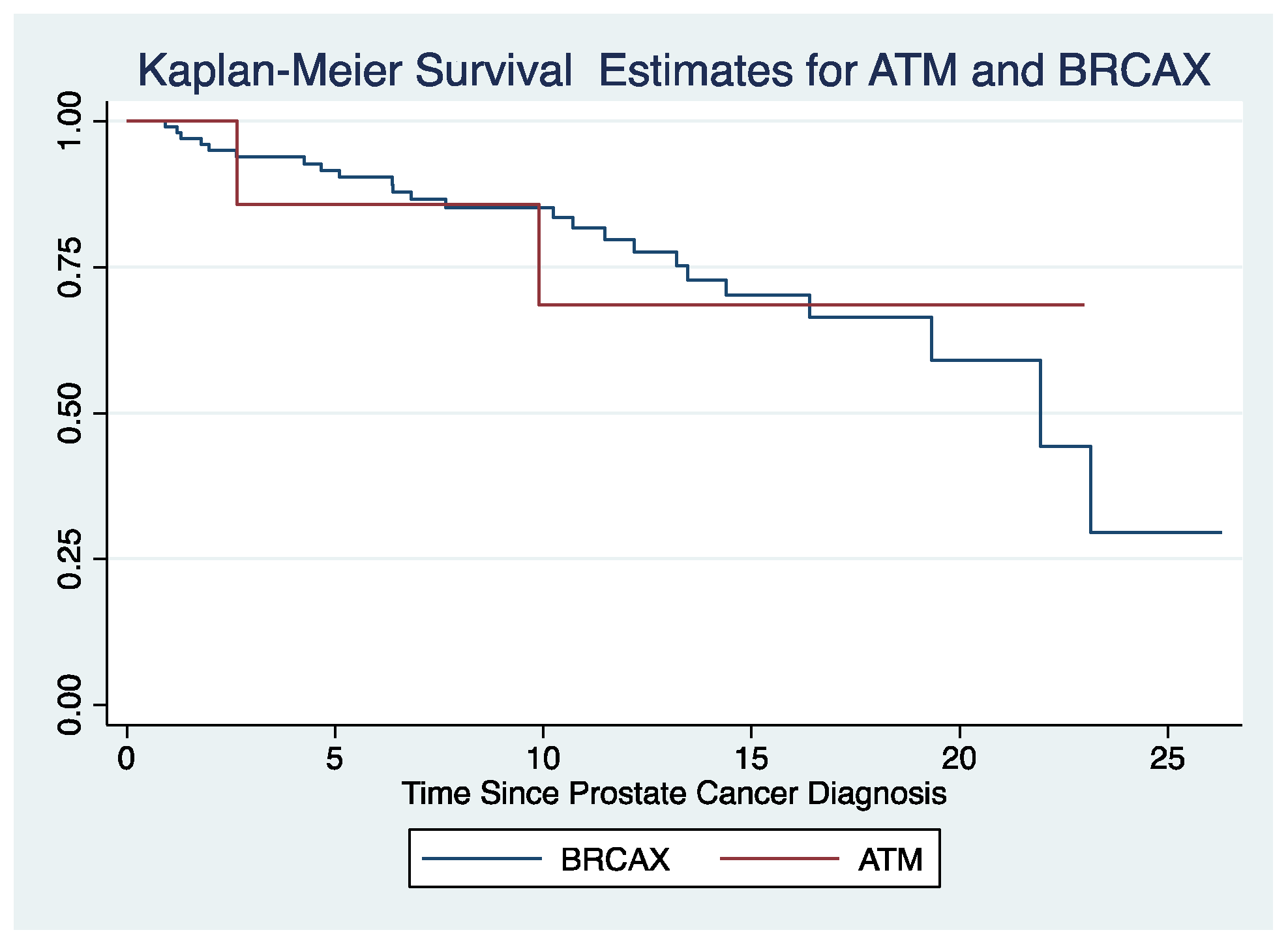
3.2. BRCAX Cohort Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thorne, H.; Willems, A.J.; Niedermayr, E.; Hoh, I.M.Y.; Li, J.; Clouston, D.; Mitchell, G.; Fox, S.; Hopper, J.L.; Bolton, D.; et al. Decreased Prostate Cancer-Specific Survival of Men with BRCA2 Mutations from Multiple Breast Cancer Families. Cancer Prev. Res. 2011, 4, 1002–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, S.M.; Rowley, S.M.; Clouston, D.; Li, J.; Lupat, R.; Krishnananthan, N.; Risbridger, G.; Taylor, R.; Bolton, D.; Campbell, I.G.; et al. Searching for candidate genes in familial BRCAX mutation carriers with prostate cancer. Urol. Oncol. 2016, 34, 120.e9–120.e16. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; McDonnell, S.K.; FitzGerald, L.M.; DeRycke, L.; Fogarty, Z.; Giles, G.G.; MacInnis, R.J.; Southey, M.C.; Nguyen-Dumont, T.; Cancel-Tassin, G.; et al. Two-stage Study of Familial Prostate Cancer by Whole-exome Sequencing and Custom Capture Identifies 10 Novel Genes Associated with the Risk of Prostate Cancer. Eur. Urol. 2021, 79, 353–361. [Google Scholar] [CrossRef]
- Clark, R.; McAlpine, K.; Fleshner, N. A Clinical Trial of Prophylactic Prostatectomy for BRCA2 Mutation Carriers: Is Now the Time? Eur. Urol. Focus 2021, 7, 506–507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. 2019, 17, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessine, M.S.; Das, S.; Park, B.; Salami, S.S.; Kaffenberger, S.D.; Kasputis, A.; Solorzano, M.; Luke, M.; Vince, R.A.; Kaye, D.R.; et al. Initial Findings from a High Genetic Risk Prostate Cancer Clinic. Urology 2021, 156, 96–103. [Google Scholar] [CrossRef]
- Scheinberg, T.; Goodwin, A.; Ip, E.; Linton, A.; Mak, B.; Smith, D.P.; Stockler, M.R.; Strach, M.C.; Tran, B.; Young, A.L.; et al. Evaluation of a Mainstream Model of Genetic Testing for Men with Prostate Cancer. JCO Oncol. Pr. 2021, 17, e204–e216. [Google Scholar] [CrossRef]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef]
- Taylor, R.; Fraser, M.; Livingstone, J.; Espiritu, S.M.G.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.-J.; Yamaguchi, T.N.; Sliwinski, A.; et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671. [Google Scholar] [CrossRef]
- Karlsson, Q.; Brook, M.N.; Dadaev, T.; Wakerell, S.; Saunders, E.J.; Muir, K.; Neal, D.E.; Giles, G.G.; MacInnis, R.J.; Thibodeau, S.N.; et al. Rare Germline Variants in ATM Predispose to Prostate Cancer: A PRACTICAL Consortium Study. Eur. Urol. Oncol. 2021, 4, 570–579. [Google Scholar] [CrossRef]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Thorne, H.; Mitchell, G.; Fox, S. kConFab: A Familial Breast Cancer Consortium Facilitating Research and Translational Oncology. J. Natl. Cancer Inst. Monogr. 2011, 2011, 79–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leenders, G.; van der Kwast, T.H.; Grignon, D.J.; Evans, A.J.; Kristiansen, G.; Kweldam, C.F.; Iczkowski, K.A. The 2019 International Society of Urological Pathology (ISUP) Consensus Conference on Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2020, 44, e87–e99. [Google Scholar] [CrossRef]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Wein, A. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.; Cheng, Y.; Willems-Jones, A.J.; Li, J.; Niedermeyr, E.; Mitchell, G.; Clouston, D.; Lawrentschuk, N.; Sliwinski, A.; Fox, S.; et al. Altered significance of D’Amico risk classification in patients with prostate cancer linked to a familial breast cancer (kConFab) cohort. Br. J. Urol. 2014, 116, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Papa, N.; O’Callaghan, M.; James, E.; Millar, J. Prostate Cancer in Australian and New Zealand Men: Patterns of Care within PCOR-ANZ 2015–2018; Monash University & Movember: Melbourne, VIC, Australia, 2021. [Google Scholar]
- Kraft, P.; Thomas, D.C. Bias and Efficiency in Family-Based Gene-Characterization Studies: Conditional, Prospective, Retrospective, and Joint Likelihoods. Am. J. Hum. Genet. 2000, 66, 1119–1131. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Dumont., T.; Dowty, J.G.; Steen, J.A.; Renault, A.L.; Hammet, F.; Mahmoodi, M.; Theys, D.; Rewse, A.; Tsimiklis, H.; Winship, I.M.; et al. Population-Based Estimates of the Age-Specific Cumulative Risk of Breast Cancer for Pathogenic Variants in CHEK2: Findings from the Australian Breast Cancer Family Registry. Cancers 2021, 13, 1378. [Google Scholar] [CrossRef]
- Curado, M.P.; Edwards, B.; Shin, H.R.; Storm, H.; Ferlay, J.; Heanue, M.; Boyle, P. Cancer Incidence in Five Continents Volume IX; IARC Press: Lyon, France, 2007. [Google Scholar]
- Vietri, M.; D’Elia, G.; Caliendo, G.; Resse, M.; Casamassimi, A.; Passariello, L.; Albanese, L.; Cioffi, M.; Molinari, A. Hereditary Prostate Cancer: Genes Related, Target Therapy and Prevention. Int. J. Mol. Sci. 2021, 22, 3753. [Google Scholar] [CrossRef]
- Brandão, A.; Paulo, P.; Maia, S.; Pinheiro, M.; Peixoto, A.; Cardoso, M.; Silva, M.P.; Santos, C.; Eeles, R.A.; Kote-Jarai, Z.; et al. The CHEK2 Variant C.349A>G Is Associated with Prostate Cancer Risk and Carriers Share a Common Ancestor. Cancers 2020, 12, 3254. [Google Scholar] [CrossRef]
- Bancroft, E.K.; Page, E.C.; Brook, M.N.; Thomas, S.; Taylor, N.; Pope, J.; McHugh, J.; Jones, A.-B.; Karlsson, Q.; Merson, S.; et al. A prospective prostate cancer screening programme for men with pathogenic variants in mismatch repair genes (IMPACT): Initial results from an international prospective study. Lancet Oncol. 2021, 22, 1618–1631. [Google Scholar] [CrossRef]
- Nguyen-Dumont, T.; Dowty, J.; MacInnis, R.; Steen, J.; Riaz, M.; Dugué, P.-A.; Renault, A.-L.; Hammet, F.; Mahmoodi, M.; Theys, D.; et al. Rare Germline Pathogenic Variants Identified by Multigene Panel Testing and the Risk of Aggressive Prostate Cancer. Cancers 2021, 13, 1495. [Google Scholar] [CrossRef] [PubMed]
- Geary, J.; Majumder, M.; Guerrini, C.; Cook-Deegan, R. Development of an Open Database of Genes Included in Hereditary Cancer Genetic Testing Panels Available From Major Sources in the US. JAMA Oncol. 2022, 8, 7639. [Google Scholar] [CrossRef]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Cheng, H.H.; Cooney, K.A.; Cookson, M.S.; Dahut, W.; Weissman, S.; Soule, H.R.; Petrylak, D.P.; et al. Implementation of Germline Testing for Prostate Cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J. Clin. Oncol. 2020, 38, 2798–2811. [Google Scholar] [CrossRef] [PubMed]
- Andoni, T.; Wiggins, J.; Robinson, R.; Charlton, R.; Sandberg, M.; Eeles, R. Half of germline pathogenic and likely pathogenic variants found on panel tests do not fulfil NHS testing criteria. Sci. Rep. 2022, 12, 2507. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.T.; Syed, J.; Nguyen, K.A.; Leapman, M.S.; Agarwal, N.; Ms, K.B.; Llor, X.; Hofstatter, E.; Shuch, B. Genetic testing for hereditary prostate cancer: Current status and limitations. Cancer 2018, 124, 3105–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Leongamornlert, D.; Collaborators, T.U.; Saunders, E.; Dadaev, T.; Tymrakiewicz, M.; Goh, C.; Jugurnauth-Little, S.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; et al. Frequent germline deleterious mutations in DNA repair genes in familial prostate cancer cases are associated with advanced disease. Br. J. Cancer 2014, 110, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- (MSAC) MSAC. 1618—Testing of Tumour Prostate Tissue to Detect BRCA1/2 Pathogenic Gene Variants in Men with Metastatic Castration-Resistant Prostate Cancer to Help Determine Eligibility for PBS Olaparib 2022. Updated 21 February 2022. Available online: http://www.msac.gov.au/internet/msac/publishing.nsf/Content/1618-public (accessed on 16 April 2022).
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline Mutations in HOXB13 and Prostate-Cancer Risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Mersch, J.; Brown, N.; Pirzadeh-Miller, S.; Mundt, E.; Cox, H.C.; Brown, K.; Aston, M.; Esterling, L.; Manley, S.; Ross, T. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA 2018, 320, 1266–1274. [Google Scholar] [CrossRef]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Nomenclature | Affected Families (n) | Carriers with PCa (n) | Non-Carriers with PCa (n) | Recruitment Stream |
|---|---|---|---|---|---|
| ATM | c.6115G > A; p.Glu2039Lys | 1 | 2 | 0 | Breast/ovarian |
| c.8266A > T; p.Lys2756Ter | 1 | 2 | 0 | Breast/ovarian | |
| c.8395_8404del | 1 | 2 | 1 | Prostate | |
| c.3712_3716del; p.Leu1238Lysfs 6 | 1 | 3 | 0 | Prostate | |
| CHEK2 | c.349A > G; p.Arg117Gly | 1 | 1 | 0 | Breast/ovarian |
| c.499G > A; p.Gly167Arg | 1 | 1 | 0 | Breast/ovarian | |
| HOXB13 | G84E | 3 | 4 | 0 | Breast/ovarian |
| PTCH1 | c.3850C > T; p.Gln1284 | 1 | 1 | 0 | Breast/ovarian |
| c.290dup; p.Asn97Lysfs 43 | 1 | 1 | 0 | Breast/ovarian | |
| BARD1 | c.1921C > T; p.Arg641 | 1 | 2 | 0 | Breast/ovarian |
| NBN | c.217A>T; p.Lys73 | 1 | 1 | 0 | Breast/ovarian |
| RECQL4 | c.2269C > T; p.Gln757 | 1 | 1 | 1 | Breast/ovarian |
| WRN | c.3030_3033del; p.Thr1011Argfs 11 | 1 | 1 | Prostate | |
| BRCA1 | c.2612insT; p.PHe872Valsfs 31 | 1 | 2 | 0 | Breast/ovarian |
| c.2864C > A; p.Ser955 | 1 | 0 | 0 | Prostate | |
| c.547+1G > T (Splice donor) | 1 | 2 | 0 | Prostate | |
| BRCA2 | c.67+1G > T; (Splice donor) | 1 | 1 | 0 | Breast/ovarian |
| c.7266T > A; p.Cys2422 | 1 | 2 | 0 | Prostate | |
| c.5909C > A; p.Ser1970 | 1 | 1 | 0 | Prostate | |
| c.8756G > A; p.Gly2919Asp | 1 | 1 | 0 | Prostate |
| ATM Mutation Carriers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at Diagnosis | PSA 1 | Grade Group | Stage | Architecture 2 | D’Amico | Primary Treatment | Age at Death | Cause of Death | Age at Diagnosis of Other Primary Malignancy |
| 78 | 13.6 | - | - | - | Int | Radiation | 87 | PCa | - |
| 90 | 210 | 5 | T1c | No | High | Non-curative | 93 | PCa | - |
| 57 | 160 | - | - | - | High | Radiation | 75 | MDS 3 | 70, MDS |
| 65 | 20 | 5 | T1c | Cribriform | High | Radiation | - | - | 66, Renal |
| 75 | 5 | 3 | T3aN1 | Cribriform | High | Prostatectomy | - | - | |
| 68 | 12 | 5 | T3b | IDCP | High | Prostatectomy | - | - | - |
| 49 | 2.6 | 3 | T2a | IDCP, Cribriform | Int | Prostatectomy | - | - | - |
| 42 | 6.9 | 1 | - | Low | Prostatectomy | - | - | - | |
| 61 | 12 | 2 | T2c | - | High | Prostatectomy | - | - | 83, Lung, Melanoma |
| CHEK2 Mutation Carriers | |||||||||
| 80 | - | 5 | T4 | - | High | Prostatectomy | 81 | Cardiac | 78, Bladder, Colon |
| 56 | 8.8 | 2 | T2c | - | High | Prostatectomy | - | - | 47, Breast |
| HOXB13G84E Mutation Carriers | |||||||||
| 68 | 6.7 | 1 | T1c | Low | Radiation | 92 | - | 80, Breast | |
| 69 | 5.3 | 2 | T2N0 | Int | Prostatectomy | - | - | - | |
| 47 | 3.8 | 2 | T3a | High | Prostatectomy | - | - | - | |
| 64 | - | 2 | T2a | Int | - | 89 | PCa | - | |
| Percentage Cumulative Risk (95% CI) of Prostate Cancer Diagnosis | ||
|---|---|---|
| Age (Years) | ATM Mutation Carriers | CHEK2 Mutation Carriers |
| 30 | 0 (0–0) | 0 (0–0.1) |
| 40 | 0 (0–0) | 0 (0–0.4) |
| 50 | 0.5 (0.1–2.5) | 1.2 (0.1–22.4) |
| 60 | 5.7 (1.1–25.9) | 13.2 (0.7–95) |
| 70 | 20.6 (4.4–69.4) | 42.8 (2.6–100) |
| 80 | 40.9 (9.7–93.3) | 72.1 (5.8–100) |
| BRCAX (n = 111) | PCOR1 (n = 39,953) | BRCA2 (n = 40) 1 | |
|---|---|---|---|
| Age at Diagnosis | 62.7 (mean) 62 (median) 42–84 (range) 9.7 (SE) | 68 (median) | 65.9 (mean) 64.95 (median) |
| PSA pre-diagnosis, n (%) | |||
| <4 ng/mL | 17 (15.3%) | 955 (13%) | 1 (2.5%) |
| 4–10 ng/mL | 43 (38.7%) | 4111 (56%) | 10 (25%) |
| 10+ ng/mL | 25 (22.5%) | 2225 (31%) | 14 (35%) |
| Unknown | 26 (23.4%) | 1067 (14%) | 11 (27.5%) |
| ISUP Grade Group at Diagnosis, n (%) | |||
| 1 | 19 (17.1%) | 2115 (26%) | 2 (5.3%) |
| 2 | 32 (28.8%) | 2606 (32%) | 11 (28.9%) |
| 3 | 23 (20.7%) | 11,380 (7%) | |
| 4 & 5 | 34 (30.6%) | 2002 (25%) | 25 (65.8%) |
| Unknown | 3 (2.7%) | 255 (4%) | - |
| D’Amico Risk Group at Diagnosis | |||
| Low | 21 (18.9%) | 1492 (20%) | 3 (7.7%) |
| Intermediate | 31 (27.9%) | 3454 (46%) | 5 (12.8%) |
| High or very high | 59 (53.2%) | 1832 (25%) | 31 (79.5%) |
| Survival Status | |||
| Deceased n (%) | 30 (27%) | - | 23 (57.5) |
| PCa-specific death (n) | 24 (21.6%) | - | 21 (52.5%) |
| Mean, median years of follow up from diagnosis (range) | 9.5, 8.6 (0.2–26.3) | – | 4.6 years (high risk)9 years (int. risk) |
| Mean, median duration to death (years) | 9.9, 8.8 (0.9–26.3) | – | 4.5 (mean) |
| PCa-specific survival estimate | 91% 5-yr (84–96) | 95% 5-yr (2012–2016) 2 | 33% 15-yr (int. risk) |
| PCa-specific survival estimate | 85% 10-yr (76–91) | 91.3% 10-yr (2011–2015) 3 | 0% 15-yr (high risk) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondschein, R.; Bolton, D.; Clouston, D.; Dowty, J.; Kavanagh, L.; Murphy, D.; Scott, P.; Taylor, R.A.; Thorne, H. Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer. Cancers 2022, 14, 3623. https://doi.org/10.3390/cancers14153623
Mondschein R, Bolton D, Clouston D, Dowty J, Kavanagh L, Murphy D, Scott P, Taylor RA, Thorne H. Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer. Cancers. 2022; 14(15):3623. https://doi.org/10.3390/cancers14153623
Chicago/Turabian StyleMondschein, Romy, Damien Bolton, David Clouston, James Dowty, Liam Kavanagh, Declan Murphy, Prudence Scott, Renea A. Taylor, and Heather Thorne. 2022. "Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer" Cancers 14, no. 15: 3623. https://doi.org/10.3390/cancers14153623
APA StyleMondschein, R., Bolton, D., Clouston, D., Dowty, J., Kavanagh, L., Murphy, D., Scott, P., Taylor, R. A., & Thorne, H. (2022). Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer. Cancers, 14(15), 3623. https://doi.org/10.3390/cancers14153623