
BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside

 ,
,  , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
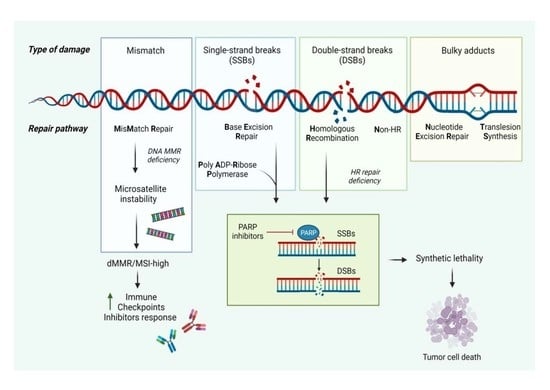
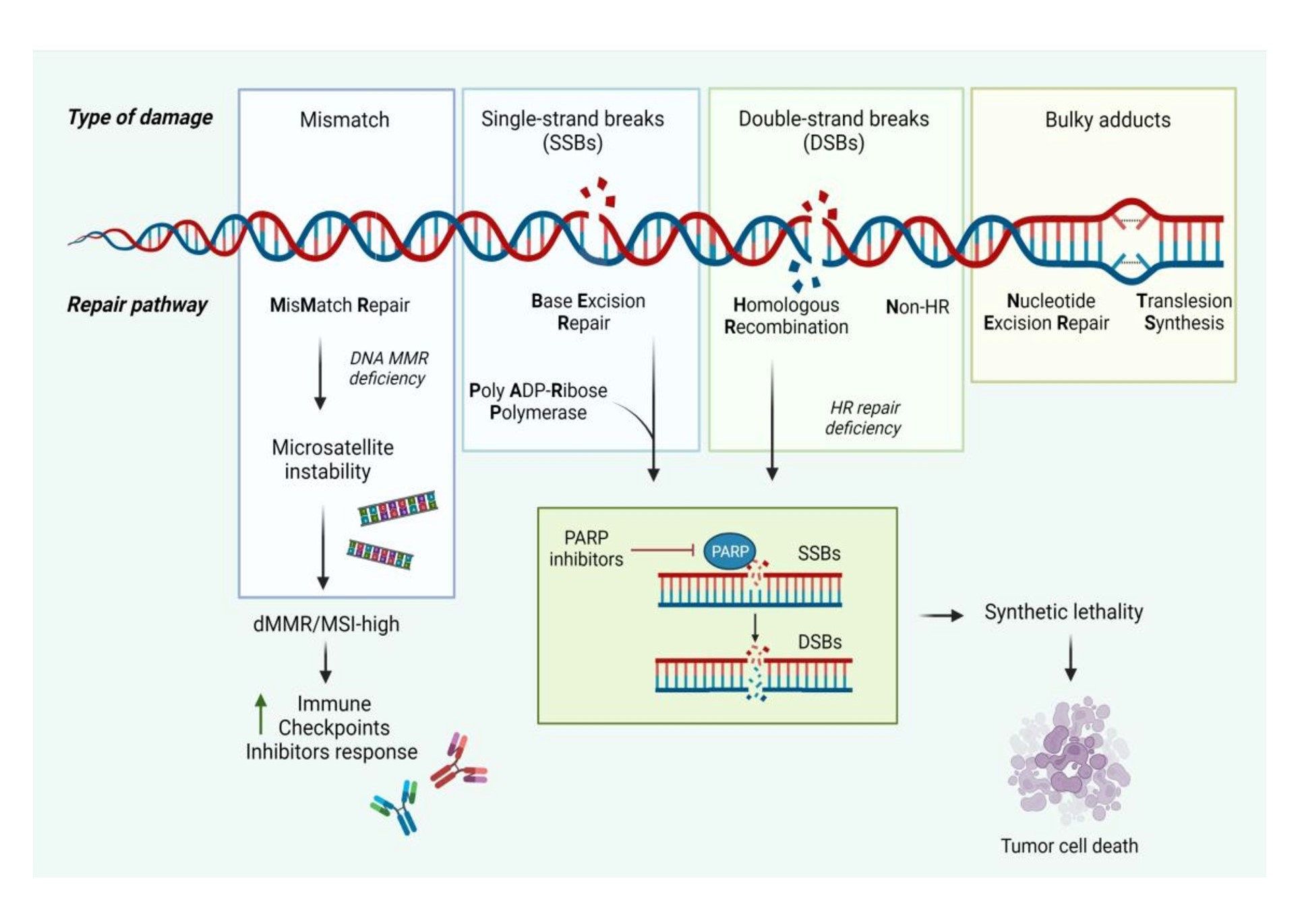
1. Introduction
2. Molecular Landscape
2.1. Homologous Recombination and Nonhomologous End Joining
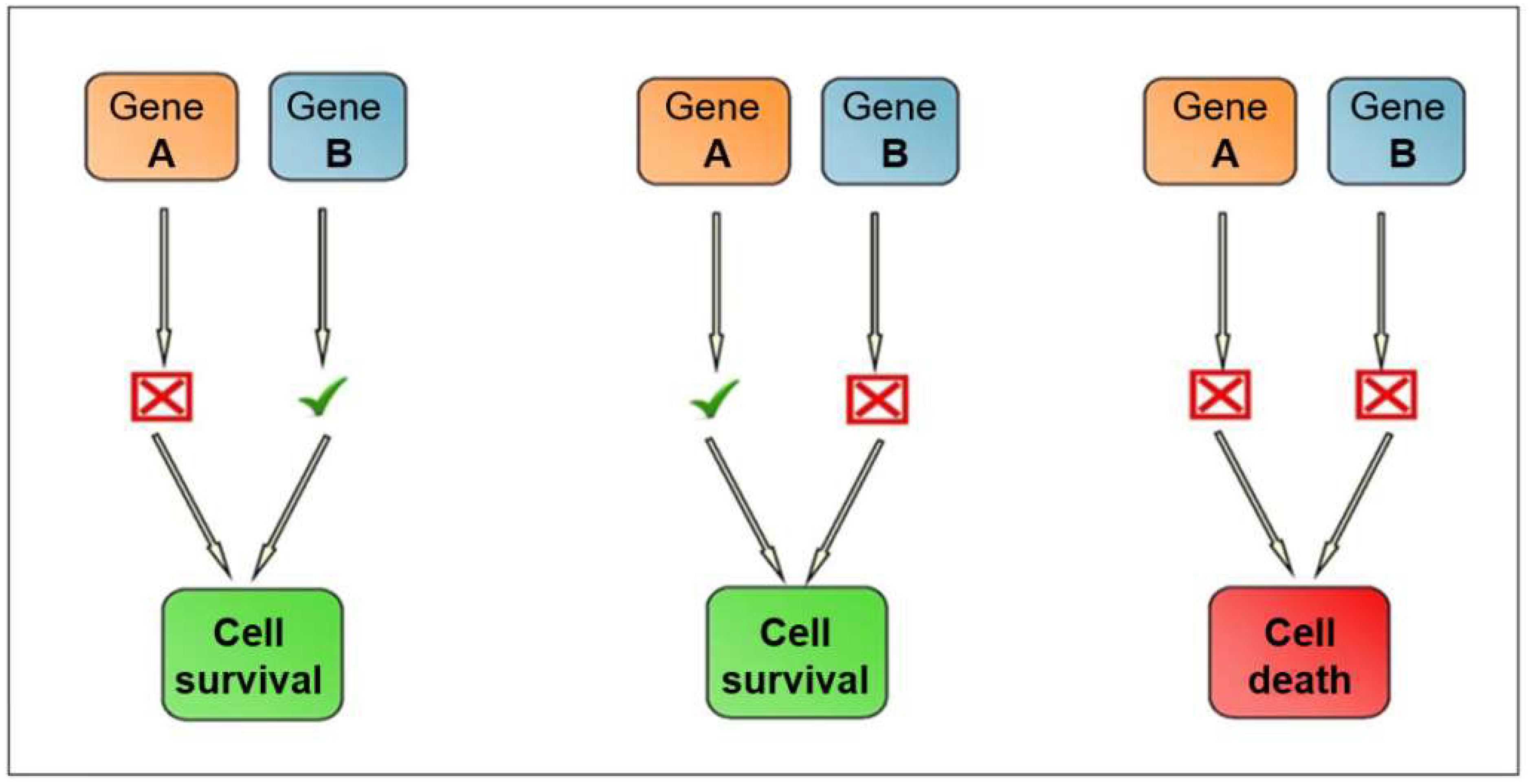
2.2. Synthetic Lethality
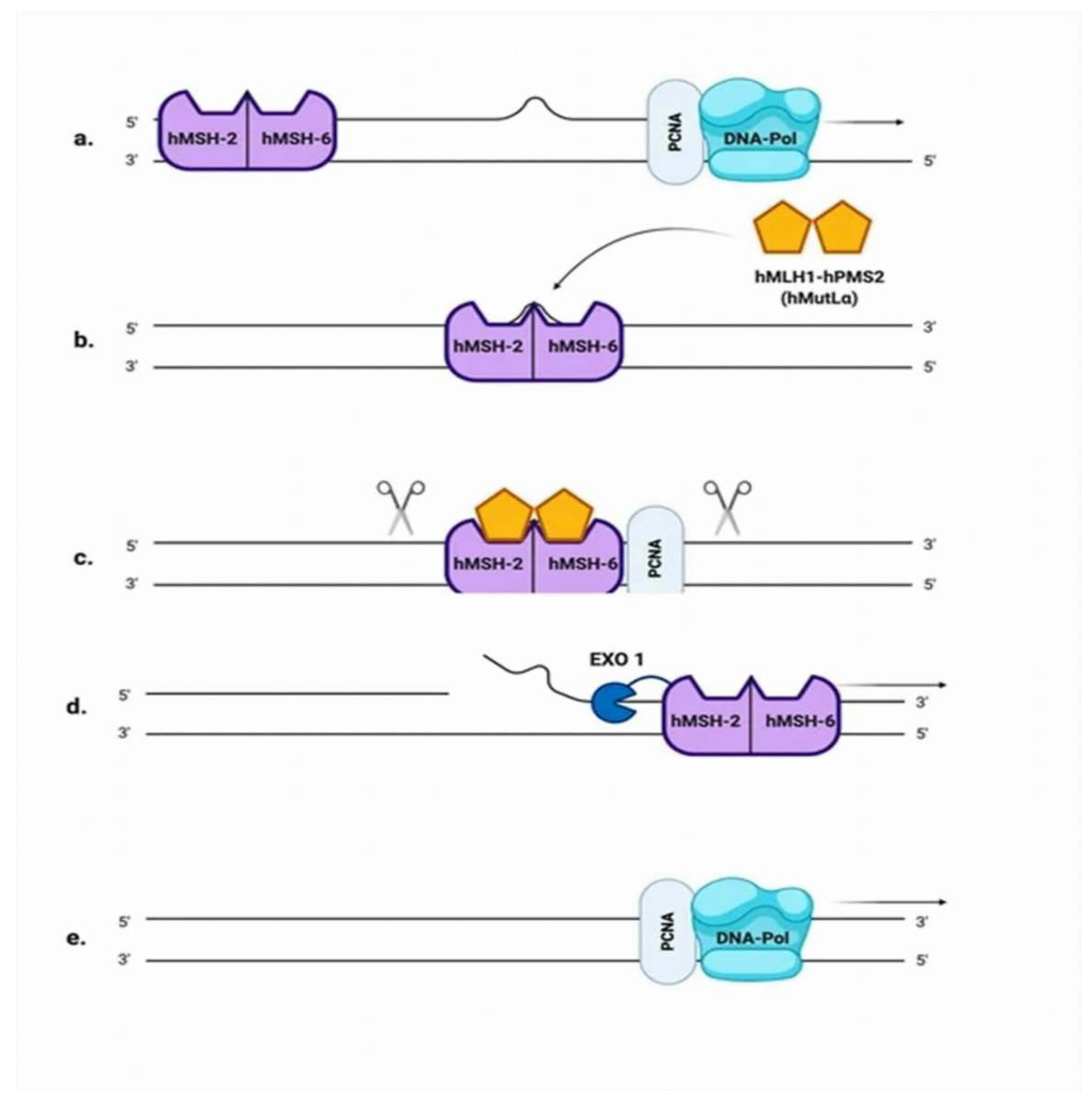
2.3. Mismatch Repair (MMR)
3. Susceptibility to EOC
3.1. BRCA1 and BRCA2 Genes in EOC
3.2. Beyond BRCA1 and BRCA2 Genes in Ovarian Cancer
3.3. Tumour Testing in Ovarian Cancer
4. Susceptibility to Prostate Cancer
4.1. BRCA1 and BRCA2 Genes in Prostate Cancer
4.2. Beyond BRCA
4.3. Tumour Testing in Prostate Cancer
5. PARP Inhibitors Development across Tumour Types
5.1. Development of PARP Inhibitors in EOC
5.2. Development of PARP Inhibitors in Prostate Cancer
6. Developing Predictive Biomarkers for PARP Inhibitors
7. BRCA Mutations and Radiation Response
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.; Moschetta, M.; Pappas-Gogos, G.; Sheriff, M.; Boussios, S. Genetic Aberrations of DNA Repair Pathways in Prostate Cancer: Translation to the Clinic. Int. J. Mol. Sci. 2021, 22, 9783. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Moschetta, M.; Karihtala, P.; Samartzis, E.P.; Sheriff, M.; Pappas-Gogos, G.; Ozturk, M.A.; Uccello, M.; Karathanasi, A.; Tringos, M.; et al. Development of new poly(ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer: Quo Vadis? Ann. Transl. Med. 2020, 8, 1706. [Google Scholar] [CrossRef]
- Revythis, A.; Limbu, A.; Mikropoulos, C.; Ghose, A.; Sanchez, E.; Sheriff, M.; Boussios, S. Recent Insights into PARP and Immuno-Checkpoint Inhibitors in Epithelial Ovarian Cancer. Int. J. Environ. Res. Public Health 2022, 19, 8577. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Yamauchi, H.; Takei, J. Management of hereditary breast and ovarian cancer. Int. J. Clin. Oncol. 2018, 23, 45–51. [Google Scholar] [CrossRef]
- Sekine, M.; Enomoto, T.; Arai, M.; Den, H.; Nomura, H.; Ikeuchi, T.; Nakamura, S.; Registration Committee of the Japanese Organization of Hereditary Breast and Ovarian Cancer. Differences in age at diagnosis of ovarian cancer for each BRCA mutation type in Japan: Optimal timing to carry out risk-reducing salpingo-oophorectomy. J. Gynecol. Oncol. 2022, 33, e46. [Google Scholar] [CrossRef]
- Pavlidis, N.; Rassy, E.; Vermorken, J.B.; Assi, T.; Kattan, J.; Boussios, S.; Smith-Gagen, J. The outcome of patients with serous papillary peritoneal cancer, fallopian tube cancer, and epithelial ovarian cancer by treatment eras: 27 years data from the SEER registry. Cancer Epidemiol. 2021, 75, 102045. [Google Scholar] [CrossRef]
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628. [Google Scholar] [CrossRef]
- Boussios, S.; Rassy, E.; Shah, S.; Ioannidou, E.; Sheriff, M.; Pavlidis, N. Aberrations of DNA repair pathways in prostate cancer: A cornerstone of precision oncology. Expert. Opin. Ther. Targets 2021, 25, 329–333. [Google Scholar] [CrossRef]
- Katsuki, Y.; Jeggo, P.A.; Uchihara, Y.; Takata, M.; Shibata, A. DNA double-strand break end resection: A critical relay point for determining the pathway of repair and signaling. Genome Instab. Dis. 2020, 1, 155–171. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: What is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef]
- Ghosh, D.; Raghavan, S.C. Nonhomologous end joining: New accessory factors fine tune the machinery. Trends Genet. 2021, 37, 582–599. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, D. Repair pathway choice for double-strand breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Zang, Y.; Pascal, L.E.; Zhou, Y.; Qiu, X.; Wei, L.; Ai, J.; Nelson, J.B.; Zhong, M.; Xue, B.; Wang, S.; et al. ELL2 regulates DNA non-homologous end joining (NHEJ) repair in prostate cancer cells. Cancer Lett. 2018, 415, 198–207. [Google Scholar] [CrossRef]
- Biswas, H.; Goto, G.; Wang, W.; Sung, P.; Sugimoto, K. Ddc2ATRIP promotes Mec1ATR activation at RPA-ssDNA tracts. PLoS Genet. 2019, 15, e1008294. [Google Scholar] [CrossRef]
- Koch, C.A.; Agyei, R.; Galicia, S.; Metalnikov, P.; O’Donnell, P.; Starostine, A.; Weinfeld, M.; Durocher, D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004, 23, 3874–3885. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs. 2020, 38, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Al-Mugotir, M.; Lovelace, J.J.; George, J.; Bessho, M.; Pal, D.; Struble, L.; Kolar, C.; Rana, S.; Natarajan, A.; Bessho, T.; et al. Selective killing of homologous recombination-deficient cancer cell lines by inhibitors of the RPA:RAD52 protein-protein interaction. PLoS ONE 2021, 16, e0248941. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids. Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Patel, P.S.; Algouneh, A.; Hakem, R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: Where we stand. Oncogene 2021, 40, 3001–3014. [Google Scholar] [CrossRef]
- Mahajan, S.; Raina, K.; Verma, S.; Rao, B.J. Human RAD52 protein regulates homologous recombination and checkpoint function in BRCA2 deficient cells. Int. J. Biochem. Cell Biol. 2019, 107, 128–139. [Google Scholar] [CrossRef]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef]
- Adamson, A.W.; Ding, Y.C.; Mendez-Dorantes, C.; Bailis, A.M.; Stark, J.M.; Neuhausen, S.L. The RAD52 S346X variant reduces risk of developing breast cancer in carriers of pathogenic germline BRCA2 mutations. Mol. Oncol. 2020, 14, 1124–1133. [Google Scholar] [CrossRef]
- Biswas, K.; Sharan, S.K. RAD52 S346X variant reduces breast cancer risk in BRCA2 mutation carriers. Mol. Oncol. 2020, 14, 1121–1123. [Google Scholar] [CrossRef]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef]
- Xiao, X.; Melton, D.W.; Gourley, C. Mismatch repair deficiency in ovarian cancer—Molecular characteristics and clinical implications. Gynecol. Oncol. 2014, 132, 506–512. [Google Scholar] [CrossRef]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef]
- Adeleke, S.; Haslam, A.; Choy, A.; Diaz-Cano, S.; Galante, J.R.; Mikropoulos, C.; Boussios, S. Microsatellite instability testing in colorectal patients with Lynch syndrome: Lessons learned from a case report and how to avoid such pitfalls. Per. Med. 2022, 19, 277–286. [Google Scholar] [CrossRef]
- Reyes, G.X.; Schmidt, T.T.; Kolodner, R.D.; Hombauer, H. New insights into the mechanism of DNA mismatch repair. Chromosoma 2015, 124, 443–462. [Google Scholar] [CrossRef]
- Kumar, C.; Piacente, S.C.; Sibert, J.; Bukata, A.R.; O’Connor, J.; Alani, E.; Surtees, J.A. Multiple factors insulate Msh2-Msh6 mismatch repair activity from defects in Msh2 domain I. J. Mol. Biol. 2011, 411, 765–780. [Google Scholar] [CrossRef]
- Sokolova, A.O.; Cheng, H.H. Genetic Testing in Prostate Cancer. Curr. Oncol. Rep. 2020, 22, 5. [Google Scholar] [CrossRef]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Zhao, C.; Li, S.; Zhao, M.; Zhu, H.; Zhu, X. Prognostic values of DNA mismatch repair genes in ovarian cancer patients treated with platinum-based chemotherapy. Arch. Gynecol. Obstet. 2018, 297, 153–159. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Waqar, S.N.; Robinson, C.; Olszanski, A.J.; Spira, A.; Hackmaster, M.; Lucas, L.; Sponton, L.; Jin, H.; Hering, U.; Cronier, D.; et al. Phase I trial of ATM inhibitor M3541 in combination with palliative radiotherapy in patients with solid tumors. Investig. New Drugs 2022, 40, 596–605. [Google Scholar] [CrossRef]
- Fuchss, T.; Graedler, U.; Schiemann, K.; Kuhn, D.; Kubas, H.; Dahmen, H.; Zimmermann, A.; Zenke, F.; Blaukat, A. Highly potent and selective ATM kinase inhibitor M4076: A clinical candidate drug with strong anti-tumor activity in combination therapies [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2019 (AACR), Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Banerjee, S.; Stewart, J.; Porta, N.; Toms, C.; Leary, A.; Lheureux, S.; Khalique, S.; Tai, J.; Attygalle, A.; Vroobel, K.; et al. ATARI trial: ATR inhibitor in combination with olaparib in gynecological cancers with ARID1A loss or no loss (ENGOT/GYN1/NCRI). Int. J. Gynecol. Cancer 2021, 31, 1471–1475. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef]
- Shah, S.; Cheung, A.; Kutka, M.; Sheriff, M.; Boussios, S. Epithelial Ovarian Cancer: Providing Evidence of Predisposition Genes. Int. J. Environ. Res. Public Health 2022, 19, 8113. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Song, H.; Cicek, M.S.; Dicks, E.; Harrington, P.; Ramus, S.J.; Cunningham, J.M.; Fridley, B.L.; Tyrer, J.P.; Alsop, J.; Jimenez-Linan, M.; et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014, 23, 4703–4709. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Gronwald, J.; Karlan, B.; Rosen, B.; Huzarski, T.; Moller, P.; Lynch, H.T.; Singer, C.F.; Senter, L.; Neuhausen, S.L.; et al. Age-specific ovarian cancer risks among women with a BRCA1 or BRCA2 mutation. Gynecol. Oncol. 2018, 150, 85–91. [Google Scholar] [CrossRef]
- Finch, A.P.; Lubinski, J.; Møller, P.; Singer, C.F.; Karlan, B.; Senter, L.; Rosen, B.; Maehle, L.; Ghadirian, P.; Cybulski, C.; et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J. Clin. Oncol. 2014, 32, 1547–1553. [Google Scholar] [CrossRef]
- Piek, J.M.; Verheijen, R.H.; Kenemans, P.; Massuger, L.F.; Bulten, H.; van Diest, P.J. BRCA1/2-related ovarian cancers are of tubal origin: A hypothesis. Gynecol. Oncol. 2003, 90, 491. [Google Scholar] [CrossRef]
- Daly, M.B.; Dresher, C.W.; Yates, M.S.; Jeter, J.M.; Karlan, B.Y.; Alberts, D.S.; Lu, K.H. Salpingectomy as a means to reduce ovarian cancer risk. Cancer Prev. Res. 2015, 8, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Eccles, D.; Evans, D.G.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef]
- Ducy, M.; Sesma-Sanz, L.; Guitton-Sert, L.; Lashgari, A.; Gao, Y.; Brahiti, N.; Rodrigue, A.; Margaillan, G.; Caron, M.C.; Côté, J.; et al. The Tumor Suppressor PALB2: Inside Out. Trends Biochem. Sci. 2019, 44, 226–240. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Mills, A.M.; Longacre, T.A. Lynch Syndrome Screening in the Gynecologic Tract: Current State of the Art. Am. J. Surg. Pathol. 2016, 40, e35–e44. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Cheung, A.; Shah, S.; Parker, J.; Soor, P.; Limbu, A.; Sheriff, M.; Boussios, S. Non-Epithelial Ovarian Cancers: How Much Do We Really Know? Int. J. Environ. Res. Public Health 2022, 19, 1106. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Tsiouris, A.K.; Chatziantoniou, A.A.; Kanellos, F.S.; Tatsi, K. Ovarian carcinosarcoma: Current developments and future perspectives. Crit. Rev. Oncol. Hematol. 2019, 134, 46–55. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef]
- Lou, E.; Vogel, R.I.; Teoh, D.; Hoostal, S.; Grad, A.; Gerber, M.; Monu, M.; Lukaszewski, T.; Deshpande, J.; Linden, M.A.; et al. Assessment of Circulating Tumor Cells as a Predictive Biomarker of Histology in Women With Suspected Ovarian Cancer. Lab. Med. 2018, 49, 134–139. [Google Scholar] [CrossRef]
- Kuhlmann, J.D.; Wimberger, P.; Bankfalvi, A.; Keller, T.; Schöler, S.; Aktas, B.; Buderath, P.; Hauch, S.; Otterbach, F.; Kimmig, R.; et al. ERCC1-positive circulating tumor cells in the blood of ovarian cancer patients as a predictive biomarker for platinum resistance. Clin. Chem. 2014, 60, 1282–1289. [Google Scholar] [CrossRef]
- Yang, F.; Tang, J.; Zhao, Z.; Zhao, C.; Xiang, Y. Circulating tumor DNA: A noninvasive biomarker for tracking ovarian cancer. Reprod. Biol. Endocrinol. 2021, 19, 178. [Google Scholar] [CrossRef]
- Oh, M.; Alkhushaym, N.; Fallatah, S.; Althagafi, A.; Aljadeed, R.; Alsowaida, Y.; Jeter, J.; Martin, J.R.; Babiker, H.M.; McBride, A.; et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate 2019, 79, 880–895. [Google Scholar] [CrossRef]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Castro, E.; Mahmud, N.; Guy, M.; Edwards, S.; O’Brien, L.; Sawyer, E.; et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br. J. Cancer 2011, 105, 1230–1234. [Google Scholar] [CrossRef]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef]
- Patel, V.L.; Busch, E.L.; Friebel, T.M.; Cronin, A.; Leslie, G.; McGuffog, L.; Adlard, J.; Agata, S.; Agnarsson, B.A.; Ahmed, M.; et al. Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res. 2020, 80, 624–638. [Google Scholar] [CrossRef]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Abida, W.; Andriole, G.L.; Bangma, C.H.; Bekelman, J.E.; Benson, M.C.; Blanco, A.; Burnett, A.; et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J. Clin. Oncol. 2018, 36, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Schayek, H.; Haugk, K.; Sun, S.; True, L.D.; Plymate, S.R.; Werner, H. Tumor suppressor BRCA1 is expressed in prostate cancer and controls insulin-like growth factor I receptor (IGF-IR) gene transcription in an androgen receptor-dependent manner. Clin. Cancer Res. 2009, 15, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, N.; Eltze, E.; Semjonow, A.; Rink, M.; Andreas, A.; Mulder, L.; Hannemann, J.; Fisch, M.; Pantel, K.; Weier, H.U.; et al. BRCA1 loss preexisting in small subpopulations of prostate cancer is associated with advanced disease and metastatic spread to lymph nodes and peripheral blood. Clin. Cancer Res. 2010, 16, 3340–3348. [Google Scholar] [CrossRef] [PubMed]
- Sigorski, D.; Iżycka-Świeszewska, E.; Bodnar, L. Poly(ADP-Ribose) Polymerase Inhibitors in Prostate Cancer: Molecular Mechanisms, and Preclinical and Clinical Data. Target. Oncol. 2020, 15, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Saxby, H.; Mikropoulos, C.; Boussios, S. An Update on the Prognostic and Predictive Serum Biomarkers in Metastatic Prostate Cancer. Diagnostics 2020, 10, 549. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Mohler, J.L.; Antonarakis, E.S. NCCN Guidelines Updates: Management of Prostate Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 583–586. [Google Scholar]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Dall’Era, M.A.; McPherson, J.D.; Gao, A.C.; DeVere White, R.W.; Gregg, J.P.; Lara, P.N., Jr. Germline and somatic DNA repair gene alterations in prostate cancer. Cancer 2020, 126, 2980–2985. [Google Scholar] [CrossRef]
- Moreno, J.G.; Gomella, L.G. Evolution of the Liquid Biopsy in Metastatic Prostate Cancer. Urology 2019, 132, 1–9. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Bancroft, E.K.; Page, E.C.; Castro, E.; Lilja, H.; Vickers, A.; Sjoberg, D.; Assel, M.; Foster, C.S.; Mitchell, G.; Drew, K.; et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: Results from the initial screening round of the IMPACT study. Eur. Urol. 2014, 66, 489–499. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Prostate Cancer Early Detection. National Comprehensive Cancer Network. Version 2.2021—14 July 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate_detection.pdf (accessed on 3 June 2022).
- Ghose, A.; Gullapalli, S.V.N.; Chohan, N.; Bolina, A.; Moschetta, M.; Rassy, E.; Boussios, S. Applications of Proteomics in Ovarian Cancer: Dawn of a New Era. Proteomes 2022, 10, 16. [Google Scholar] [CrossRef]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic. Acids. Res. 2014, 42, 7762–7775. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Yap, T.A.; Im, S.A.; Schram, A.M.; Sharp, A.; Balmana, J.; Baird, R.D.; Brown, J.S.; Schwaederle, M.; Pilling, E.A.; Moorthy, G.; et al. PETRA: First in class, first in human trial of the next generation PARP1-selective inhibitor AZD5305 in patients with BRCA1/2, PALB2, or RAD51C/D mutations. Cancer Res. 2022, 82, CT007. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs R D 2020, 20, 55–73. [Google Scholar] [CrossRef]
- Demircan, N.C.; Boussios, S.; Tasci, T.; Öztürk, M.A. Current and future immunotherapy approaches in ovarian cancer. Ann. Transl. Med. 2020, 8, 1714. [Google Scholar] [CrossRef]
- Moschetta, M.; Boussios, S.; Rassy, E.; Samartzis, E.P.; Funingana, G.; Uccello, M. Neoadjuvant treatment for newly diagnosed advanced ovarian cancer: Where do we stand and where are we going? Ann. Transl. Med. 2020, 8, 1710. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Coleman, R.L.; Swisher, E.M.; Oza, A.M.; Scott, C.L.; Giordano, H.; Lin, K.K.; Konecny, G.E.; Tinker, A.; O’Malley, D.M.; Kristeleit, R.S.; et al. Refinement of prespecified cutoff for genomic loss of heterozygosity (LOH) in ARIEL2 part 1: A phase II study of rucaparib in patients (PTS) with high grade ovarian carcinoma (HGOC). J. Clin. Oncol. 2016, 34, 5540. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Luo, J.; Ou, S.; Wei, H.; Qin, X.; Jiang, Q. Comparative Efficacy and Safety of Poly (ADP-Ribose) Polymerase Inhibitors in Patients with Ovarian Cancer: A Systematic Review and Network Meta-Analysis. Front. Oncol. 2022, 12, 815265. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Saxby, H.; Boussios, S.; Mikropoulos, C. Androgen Receptor Gene Pathway Upregulation and Radiation Resistance in Oligometastatic Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 4786. [Google Scholar] [CrossRef]
- Attard, G.; Murphy, L.; Clarke, N.W.; Cross, W.; Jones, R.J.; Parker, C.C.; Gillessen, S.; Cook, A.; Brawley, C.; Amos, C.L.; et al. Abiraterone acetate and prednisolone with or without enzalutamide for high-risk non-metastatic prostate cancer: A meta-analysis of primary results from two randomised controlled phase 3 trials of the STAMPEDE platform protocol. Lancet 2022, 399, 447–460. [Google Scholar] [CrossRef]
- Knudsen, K.E. The AR-DNA repair axis: Insights into prostate cancer aggressiveness. Can. J. Urol. 2019, 26, 22–23. [Google Scholar]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Heller, G.; Ryan, C.J.; VanderWeele, D.J.; Lewis, L.D.; Tan, A.; Watt, C.; Chen, R.C.; Kohli, M.; Barata, P.C.; et al. Alliance A031902 (CASPAR): A randomized, phase (ph) 3 trial of enzalutamide with rucaparib/placebo as novel therapy in first-line metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, TPS194. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Fizazi, K.; Mateo, J.; Matsubara, N.; Shore, N.D.; Chakrabarti, J.; Chen, H.-C.; Lanzalone, S.; Niyazov, A.; et al. Talapro-3: A phase 3, double-blind, randomized study of enzalutamide (ENZA) plus talazoparib (TALA) versus placebo plus enza in patients with DDR gene mutated metastatic castration-sensitive prostate cancer (mCSPC). J. Clin. Oncol. 2022, 40, TPS221. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef]
- Gunderson, C.C.; Moore, K.N. BRACAnalysis CDx as a companion diagnostic tool for Lynparza. Expert Rev. Mol. Diagn. 2015, 15, 1111–1116. [Google Scholar] [CrossRef]
- Myriad. Tumor BRACAnalysis CDxTM. Available online: http://myriadgenetics.eu/products/tumor-bracanalysis-cdx-2/?lang=gb (accessed on 3 June 2022).
- Ohmoto, A.; Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 2017, 10, 5195–5208. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef]
- Markowski, M.C.; Antonarakis, E.S. BRCA1 Versus BRCA2 and PARP Inhibitor Sensitivity in Prostate Cancer: More Different Than Alike? J. Clin. Oncol. 2020, 38, 3735–3739. [Google Scholar] [CrossRef]
- Lotan, T.L.; Kaur, H.B.; Salles, D.C.; Murali, S.; Schaeffer, E.M.; Lanchbury, J.S.; Isaacs, W.B.; Brown, R.; Richardson, A.L.; Cussenot, O.; et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod. Pathol. 2021, 34, 1185–1193. [Google Scholar] [CrossRef]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Mandigo, A.C.; Gordon, N.; Huang, F.; Gaur, S.; de Leeuw, R.; Zhao, S.G.; Evans, J.; Han, S.; Parsons, T.; et al. PARP-1 regulates DNA repair factor availability. EMBO Mol. Med. 2018, 10, e8816. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Kan, C.; Zhang, J. BRCA1 Mutation: A Predictive Marker for Radiation Therapy? Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 281–293. [Google Scholar] [CrossRef]
- Vallard, A.; Magné, N.; Guy, J.B.; Espenel, S.; Rancoule, C.; Diao, P.; Deutsch, E.; Rivera, S.; Chargari, C. Is breast-conserving therapy adequate in BRCA 1/2 mutation carriers? The radiation oncologist’s point of view. Br. J. Radiol. 2019, 92, 20170657. [Google Scholar] [CrossRef]
- Césaire, M.; Thariat, J.; Candéias, S.M.; Stefan, D.; Saintigny, Y.; Chevalier, F. Combining PARP inhibition, radiation, and immunotherapy: A possible strategy to improve the treatment of cancer? Int. J. Mol. Sci. 2018, 19, 3793. [Google Scholar] [CrossRef]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef]
- Chargari, C.; Levy, A.; Paoletti, X.; Soria, J.C.; Massard, C.; Weichselbaum, R.R.; Deutsch, E. Methodological Development of Combination Drug and Radiotherapy in Basic and Clinical Research. Clin. Cancer Res. 2020, 26, 4723–4736. [Google Scholar] [CrossRef]
- Schneider, G.; Schmidt-Supprian, M.; Rad, R.; Saur, D. Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef]
- Martin, S.A.; Hewish, M.; Lord, C.J.; Ashworth, A. Genomic instability and the selection of treatments for cancer. J. Pathol. 2010, 220, 281–289. [Google Scholar] [CrossRef]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.L.; Peters, T.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 416. [Google Scholar] [CrossRef]
- Yi, K.; Ju, Y.S. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Mirzaei, G.; Petreaca, R.C. Distribution of copy number variations and rearrangement endpoints in human cancers with a review of literature. Mutat. Res. 2022, 824, 111773. [Google Scholar] [CrossRef]
- Mirzaei, G. GraphChrom: A Novel Graph-Based Framework for Cancer Classification Using Chromosomal Rearrangement Endpoints. Cancers 2022, 14, 3060. [Google Scholar] [CrossRef]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef]
- Polansky, H.; Schwab, H. How latent viruses cause breast cancer: An explanation based on the microcompetition model. Bosn. J. Basic Med. Sci. 2019, 19, 221–226. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
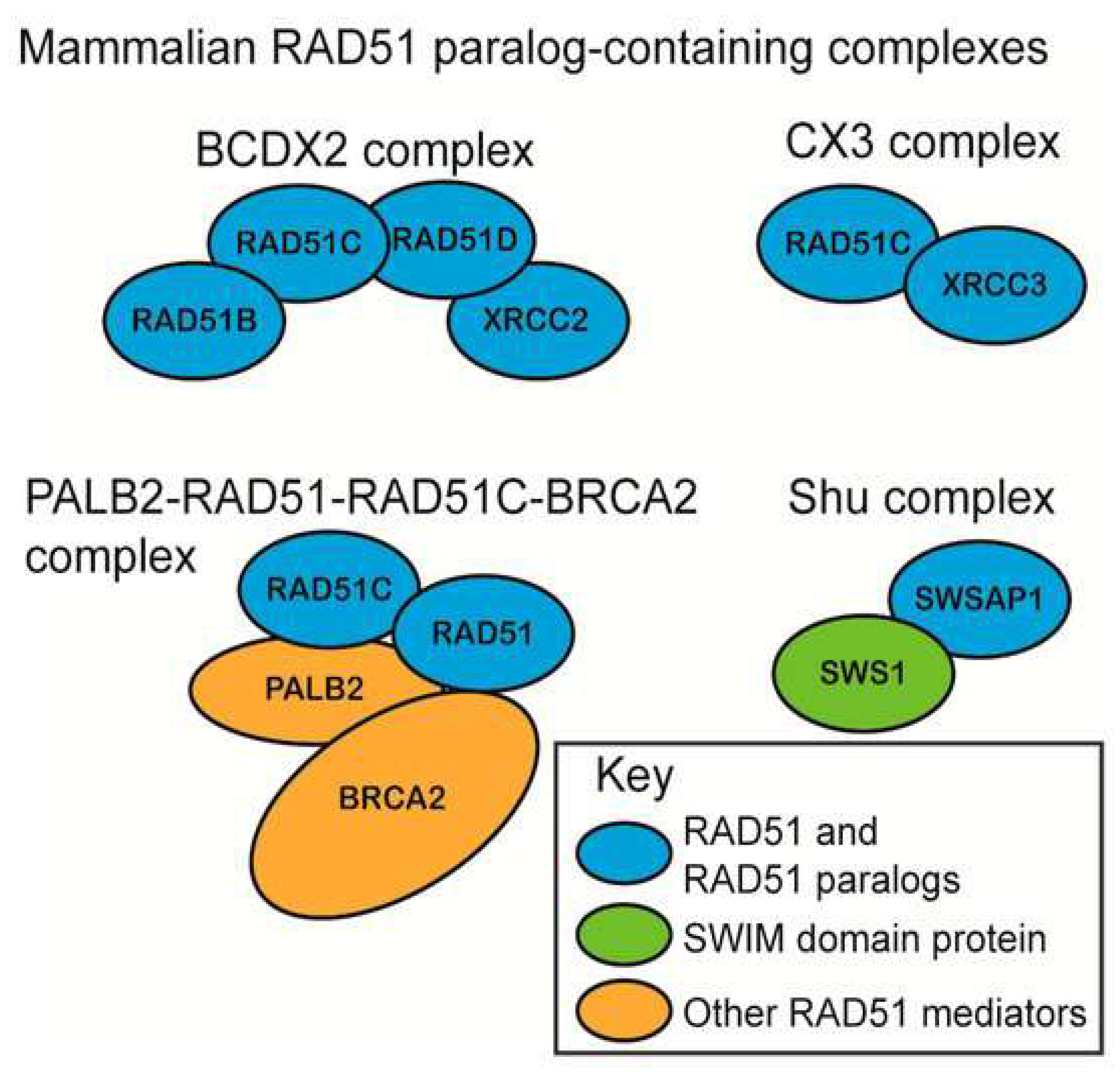{kind=link}
| Study | Phase | Population | Treatment Arms | Outcome | P | Ref |
|---|---|---|---|---|---|---|
| STUDY 19 | II | (1) Platinum-sensitive, advanced HGSOC (2) At least two prior lines of platinum-based CTH (3) Unselected for BRCA status | (A) Olaparib 400 mg BID (B) Placebo | (A): Median PFS 1. Overall population: 8.4 vs. 4.8 m 2. BRCA mutants: 11.2 vs. 4.3 m 3. BRCA wild type: 7.4 vs. 5.5 m (B): OS 1. Overall population: 29.8 vs. 27.8 m 2. BRCA mutants: 34.9 vs. 31.9 m 3. BRCA wild type: 24.5 vs. 26.2 m (C): ORR 12% vs. 4% | (A1): <0.001 (A2): <0.0001 (A3): 0.0075 (B1): 0.44 (B2): 0.19 (B3): 0.96 (C): 0.12 | [112] |
| STUDY 42 | II | (1) Platinum-resistant, advanced HGSOC (2) BRCA mutations | Olaparib 400 mg BID | (1) ORR: 34% (2) MDR: 7.9 m (3) PFS: 7 m (4) OS: 16.6 m | [113] | |
| SOLO 2 | III | (1) Platinum-sensitive, advanced HGSOC or HGEOC (2) At least two prior lines of platinum-based CTH (3) BRCA mutations | (A) Olaparib 300 mg BID (B) Placebo | Median PFS: 19.1 vs. 5.5 m | <0.0001 | [114] |
| ARIEL2 | II | Platinum-sensitive, advanced HGSOC or HGEOC | Rucaparib 600 mg BID | (A): Median PFS 1. BRCA mutants: 12.8 m 2. BRCA wild type LOH high: 5.7 m 3. BRCA wild type LOH low: 5.2 m (B): ORR 1. BRCA mutants: 80% 2. BRCA wild type LOH high: 39% 3. BRCA wild type LOH low: 13% | (A1): <0.0001 (A2): 0.011 (A3): 0.011 | [115] |
| NOVA | III | (1) Platinum-sensitive, advanced HGSOC (2) At least two prior lines of platinum-based CTH (3) Stratification by gBRCAmut | (A) Niraparib 300 mg BID (B) Placebo | Median PFS (1) gBRCA mutants: 21 vs. 5.5 m (2) BRCA wild type HRD (+): 12.9 vs. 3.8 m (3) Overall non-gBRCA mutants: 9.3 vs. 3.9 m | (1): <0.0001 (2): <0.00001 (3): <0.0001 | [117] |
| STUDY 10 | I/II | (1) Platinum-sensitive, advanced HGSOC or HGEOC; (2) gBRCAmut (phase II PART 2A) | Rucaparib 600 mg BID | (1) ORR: 59.5% (2) MDR: 7.8 m | [119] | |
| SOLO 1 | III | (1) Platinum-sensitive, advanced HGSOC (2) BRCA mutations | (A) Olaparib 300 mg BID (B) Placebo | Median PFS: NR vs. 13.8 m 3-year PFS: 69% vs. 35% | <0.001 <0.001 | [120] |
| SOLO 3 | III | Recurrent gBRCAm EOC | (A) Olaparib (B) CTH | Median PFS: 13.4 vs. 9.2 m | 0.013 | [121] |
| PRIMA | III | Newly diagnosed advanced EOC with response to platinum-based CTH | (A) Niraparib 300 mg BID (B) Placebo | Median PFS (1) HRD (+): 21.9 vs. 10.4 m (2) Overall population: 13.8 vs. 8.2 m | (1): <0.001 (2): <0.001 | [122] |
| QUADRA | II | (1) Platinum-sensitive, advanced HGSOC (2) HRD (+) | Niraparib 300 mg BID | (1) ORR 27.5% (2) DCR 68.6% | [123] | |
| ARIEL3 | III | Recurrent EOC after response to platinum-based CTH | (A) Rucaparib 600 mg BID (B) Placebo | Median PFS (1) BRCA mutants: 16.6 vs. 5.4 m (2) HRD (+): 13.6 vs. 5.4 m (3) ITT population: 10.8 vs. 5.4 m | (1): <0.0001 (2): <0.0001 (3): <0.001 | [124] |
| PAOLA-1 | III | Newly diagnosed, advanced, high-grade ovarian cancer with response after first-line platinum-taxane CTH plus bevacizumab | (A) Bevacizumab + olaparib maintenance (B) Bevacizumab + placebo | Median PFS (1) Overall population: 22.1 vs. 16.6 m (2) HRD (+): 37.2 vs. 17.7 m (3) HRD without BRCA mutations: 28.1 vs. 16.6 m | (1): <0.001 | [125] |
| Clinical Trial ID | Phase | PARP Inhibitor | Population | PSA Response Rate | Primary Endpoint | Ref |
|---|---|---|---|---|---|---|
| NCT01682772 | II | Olaparib | mCRPC patients previously treated with abiraterone or enzalutamide, and cabazitaxel | 33% of patients (95%, 20–48) | RR, PSA, CTC | [130] |
| NCT01682772 | II | Olaparib | mCRPC patients: (1) previously treated with one or two taxanes (2) DDR gene mutations | PSA levels decrease by ≥ 50%: 100% of BRCA2 and FANCA mutated mCRPC patients | RR, PSA, CTC | [131] |
| NCT02987543 | III | Olaparib | mCRPC patients: (1) disease progression whilst on enzalutamide or abiraterone (2) ≥1 HRR gene mutation | Olaparib group: 30% of patients Control group: 10% of patients | rPFS | [132] |
| NCT02952534 | II | Rucaparib | mCRPC patients: germline or somatic alteration in ≥1 prespecified HRR gene | 47.8% of BRCA-mutated patients (95%, 26.8–69.4) | ORR | [133] |
| NCT04455750 | III | Rucaparib | mCRPC patients, resistant to testosterone-deprivation therapy | Not completed | rPFS, OS | [134] |
| NCT02854436 | II | Niraparib | mCRPC patients: (1) DDR gene mutations (2) disease progression on taxane and AR-targeted therapy | 57% of patients (95% CI, 34–77) | ORR | [135] |
| NCT03148795 | II | Talazoparib | mCRPC patients: (1) DDR-mutated (2) disease progression on taxane or AR-targeted therapy | Not completed | ORR | [136] |
| NCT04821622 | III | Talazoparib | mCSPC patients with DDR gene mutations | Not completed | rPFS | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boussios, S.; Rassy, E.; Moschetta, M.; Ghose, A.; Adeleke, S.; Sanchez, E.; Sheriff, M.; Chargari, C.; Pavlidis, N. BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside. Cancers 2022, 14, 3888. https://doi.org/10.3390/cancers14163888
Boussios S, Rassy E, Moschetta M, Ghose A, Adeleke S, Sanchez E, Sheriff M, Chargari C, Pavlidis N. BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside. Cancers. 2022; 14(16):3888. https://doi.org/10.3390/cancers14163888
Chicago/Turabian StyleBoussios, Stergios, Elie Rassy, Michele Moschetta, Aruni Ghose, Sola Adeleke, Elisabet Sanchez, Matin Sheriff, Cyrus Chargari, and Nicholas Pavlidis. 2022. "BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside" Cancers 14, no. 16: 3888. https://doi.org/10.3390/cancers14163888
APA StyleBoussios, S., Rassy, E., Moschetta, M., Ghose, A., Adeleke, S., Sanchez, E., Sheriff, M., Chargari, C., & Pavlidis, N. (2022). BRCA Mutations in Ovarian and Prostate Cancer: Bench to Bedside. Cancers, 14(16), 3888. https://doi.org/10.3390/cancers14163888