Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Companion Canine Recruitment and Characteristics
2.2. Microarray Hybridization
2.3. Microarray Data Analysis
2.4. Cell Lines, Drug Selection, Methods and Materials
3. Results
3.1. Companion Canines with Non-Hodgkin-like Lymphoma Are Strong Models of MDR Malignancy
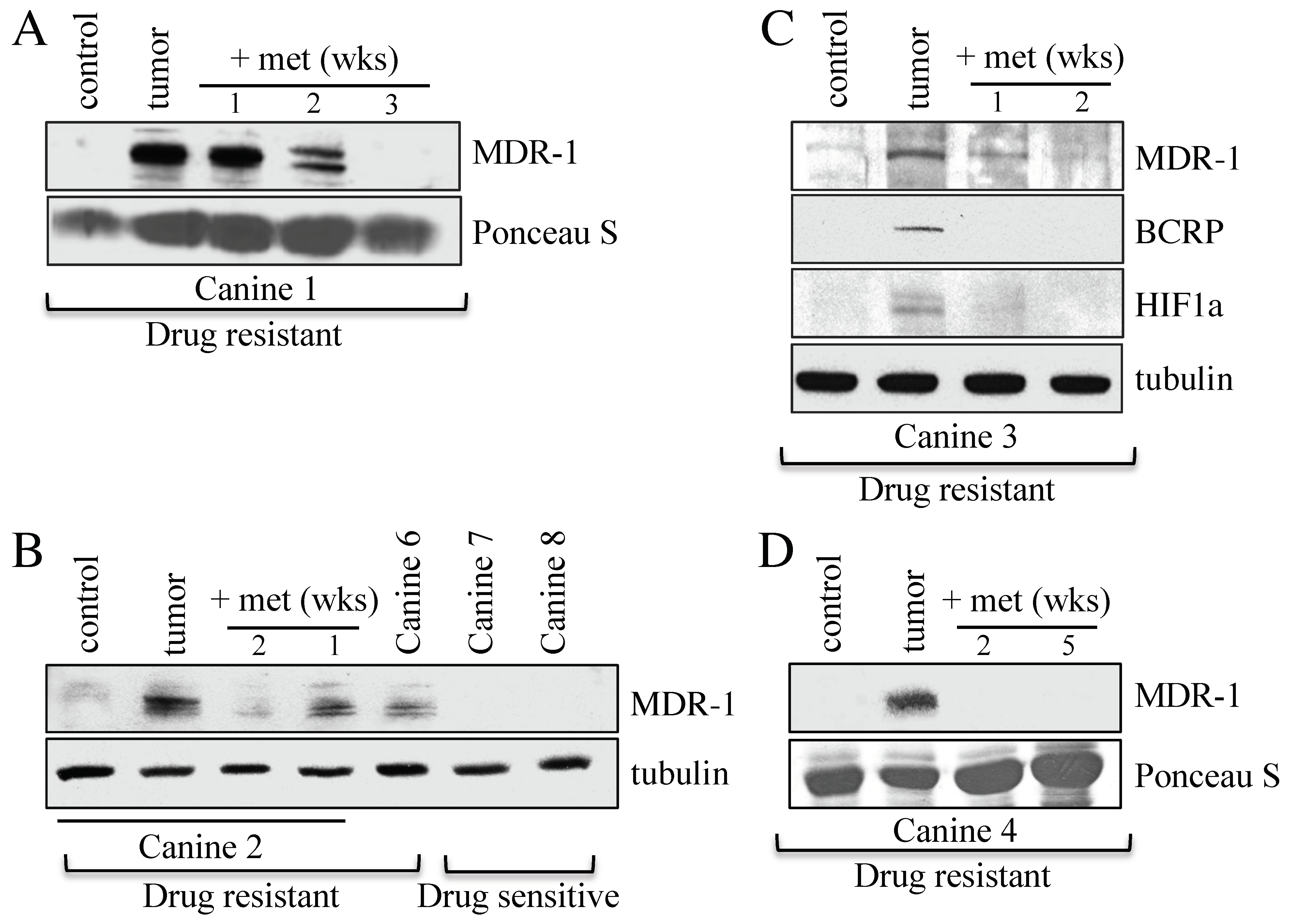
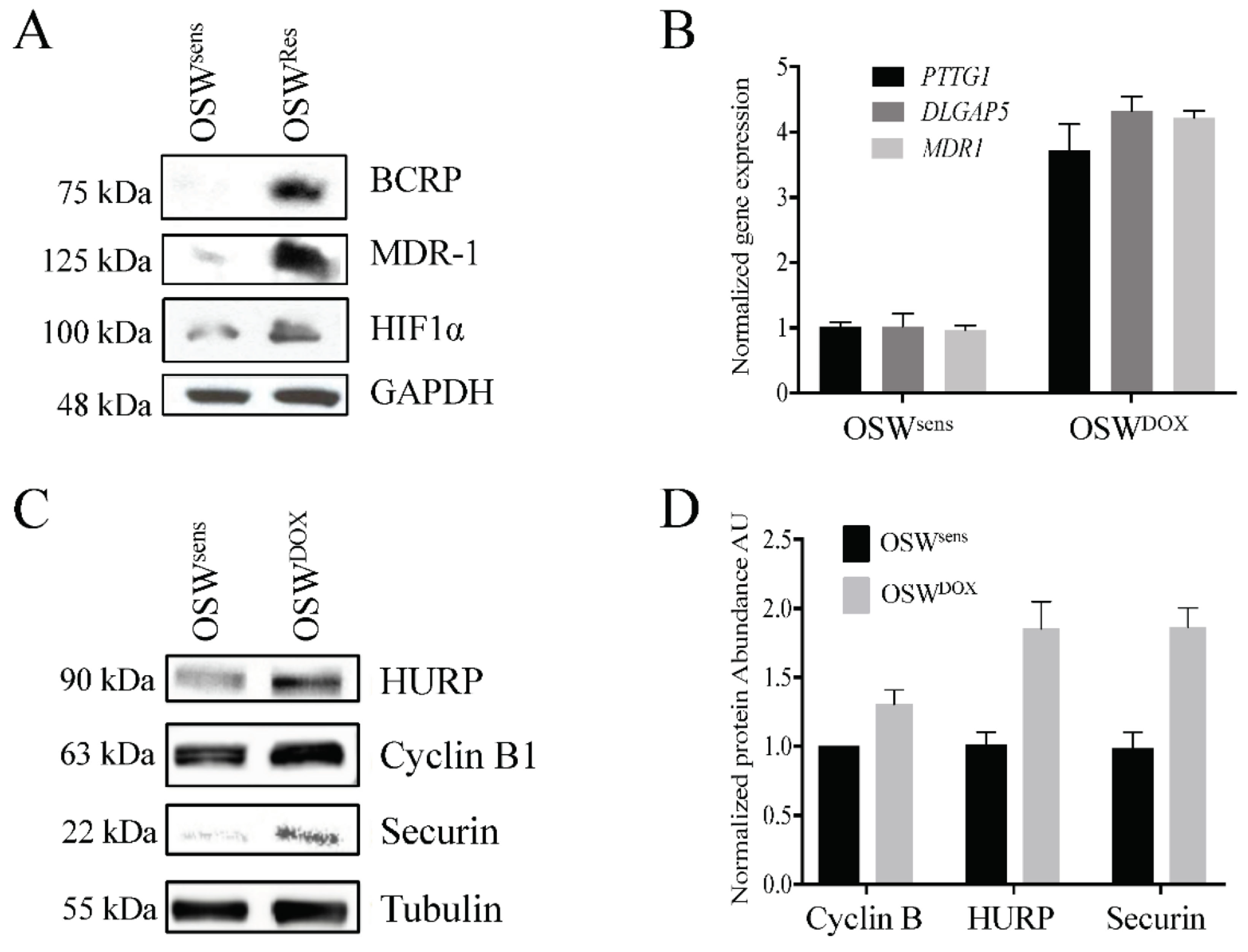
3.2. Canines with MDR Lymphoma Overexpress Proteins Associated with Drug Resistance, and Adjunct Metformin Therapy Reverses This
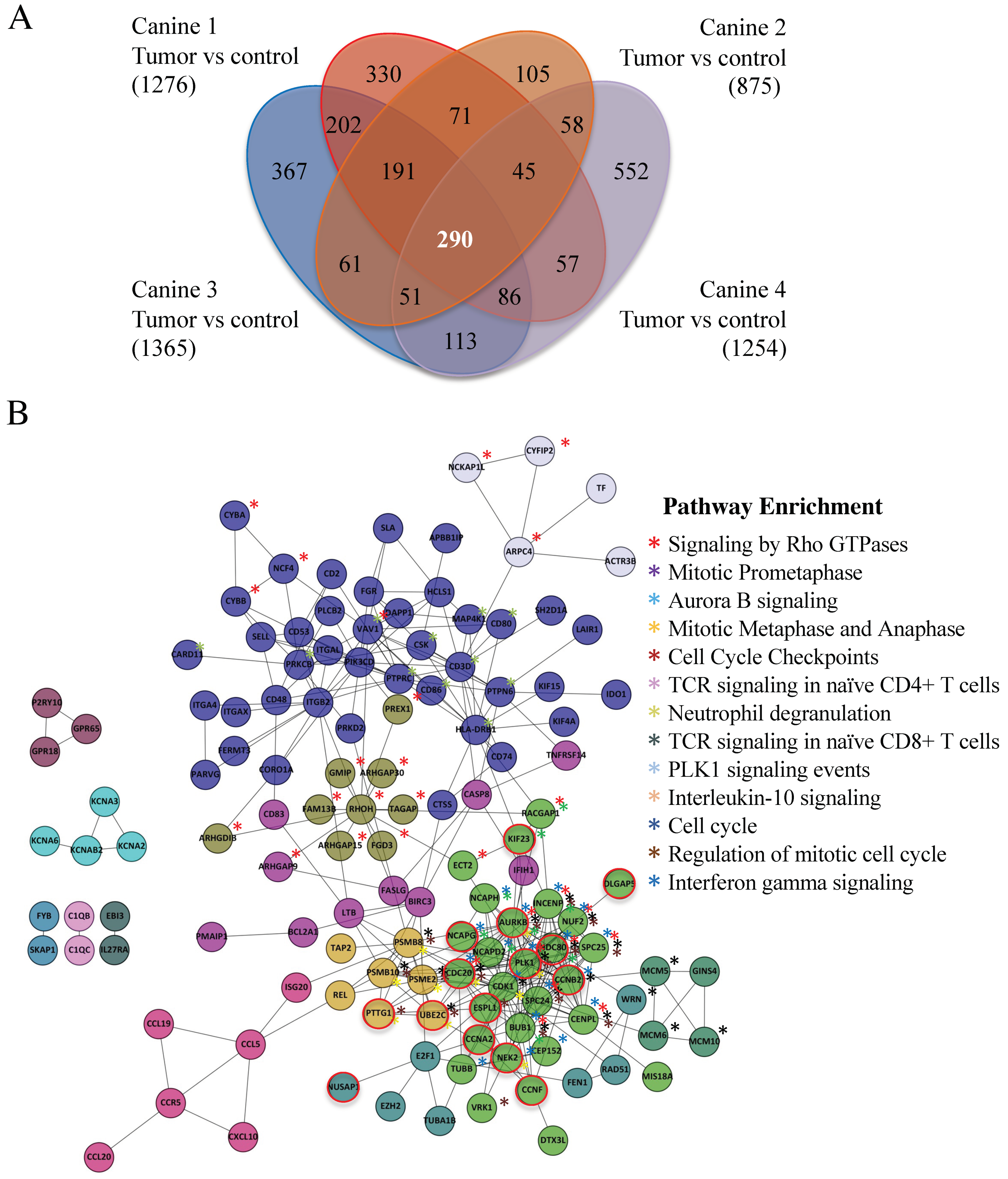
3.3. A 290 Gene Set Overexpressed > Three-Fold in Tumor Versus Control Was Common to the Four MDR Canines and Contained 20 Anaphase Promoting Complex Substrates
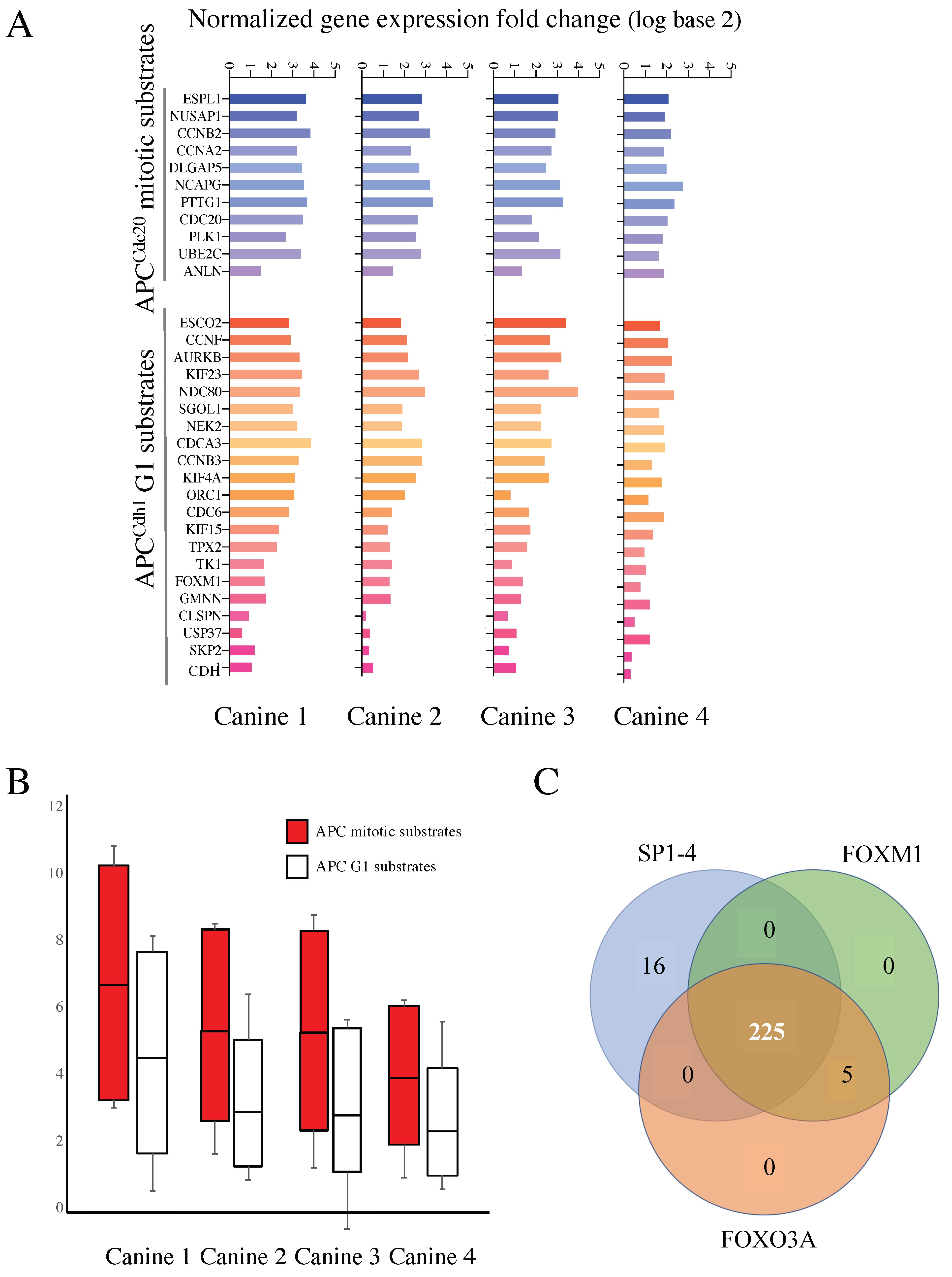
3.4. Mitotic and G1 APC Substrate Gene Expression Is Elevated in Treatment Resistant Tumors Compared to Normal Controls
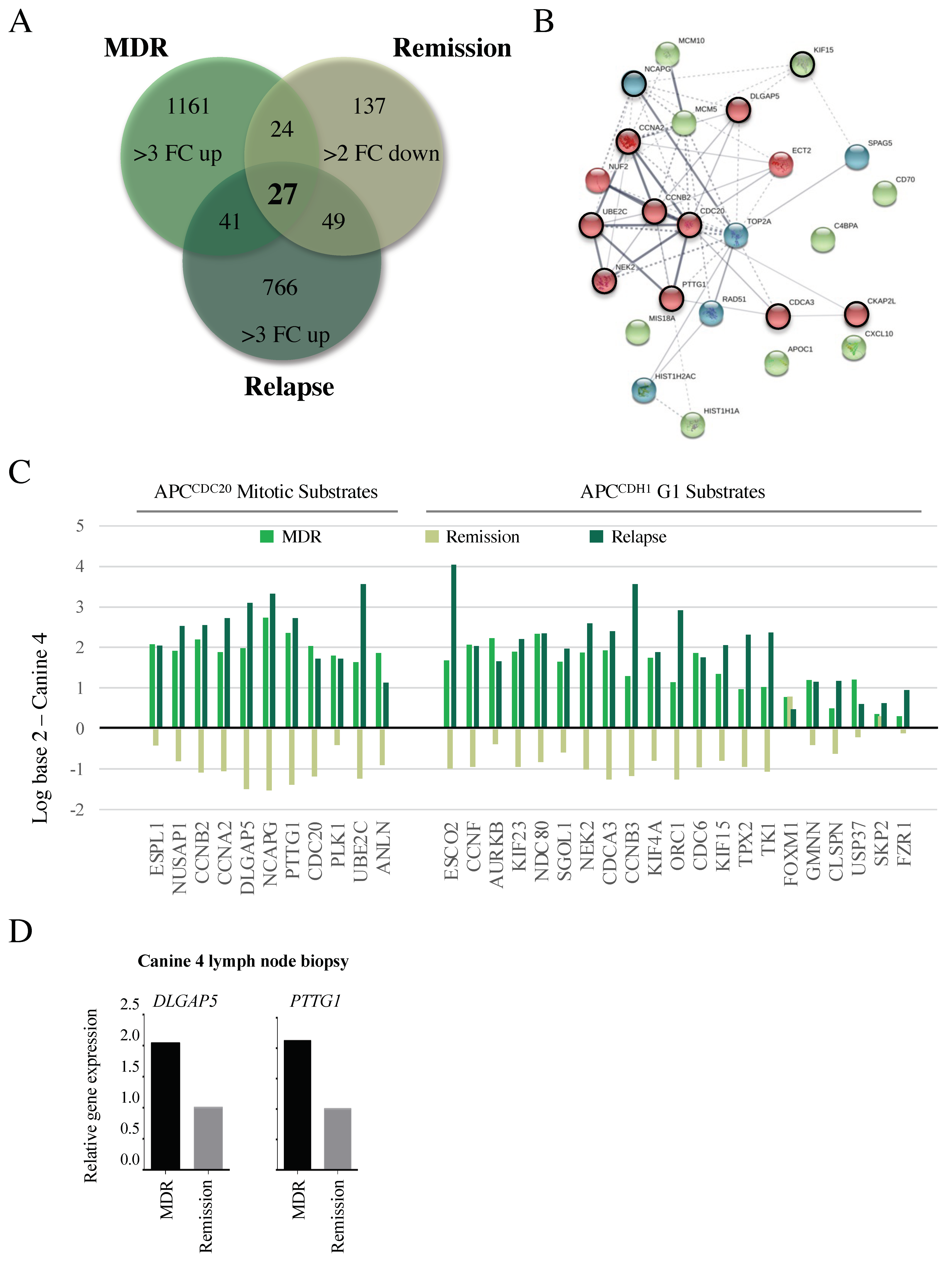
3.5. Microarray Reveals Reversible Changes in APC Target Gene Expression That Correlate with Altered Clinical Treatment Responses
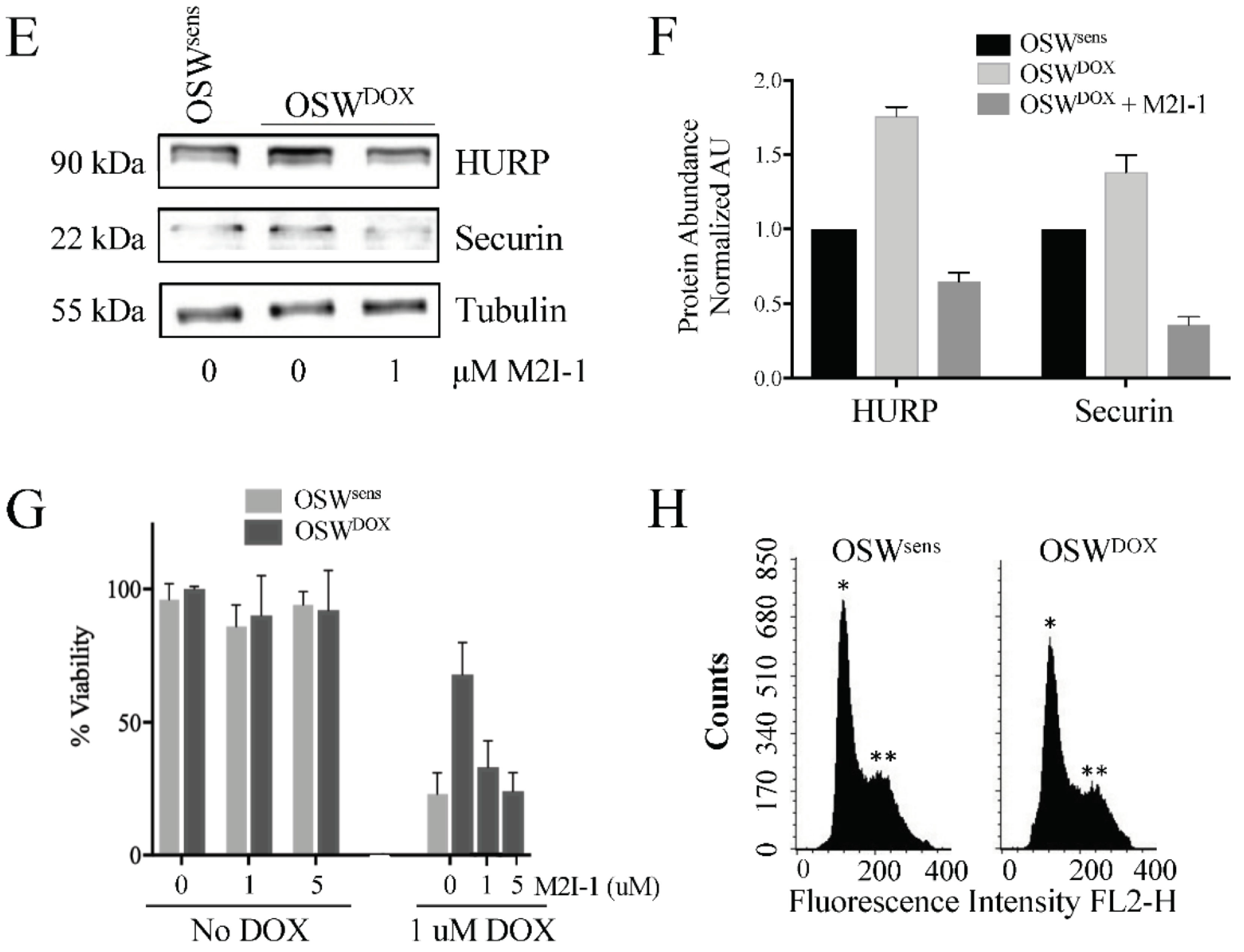
3.6. APC Substrates Are Elevated In Vitro in OSW Lymphoma Cells Selected for DOX Resistance and Reversed When the APC Is Activated
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-H.; Park, J.H.; Fan, S. Predicting and Overcoming Chemotherapeutic Resistance in Breast Cancer. Transl. Res. Breast Cancer 2017, 1026, 59–104. [Google Scholar] [CrossRef]
- Reddy, S.M.; Barcenas, C.H.; Sinha, A.K.; Hsu, L.; Moulder, S.L.; Tripathy, D.; Hortobagyi, G.N.; Valero, V. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease free at 5 years and relationship with low hormone receptor positivity. Br. J. Cancer 2017, 118, 17–23. [Google Scholar] [CrossRef]
- Arnason, T.; Harkness, T. Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1α. Cancers 2015, 7, 2063–2082. [Google Scholar] [CrossRef]
- Davies, G.F.; Roesler, W.J.; Juurlink, B.H.; Harkness, T.A. Troglitazone overcomes doxorubicin-resistance in resistant K562 leu-kemia cells. Leuk. Lymphoma 2005, 46, 1199–1206. [Google Scholar] [CrossRef]
- Harkness, T.A.; Davies, G.F.; Juurlink, B.H. Troglitazone reverses the multiple drug resistance phenotype in cancer cells. Drug Des. Dev. Ther. 2009, 3, 79–88. [Google Scholar] [CrossRef]
- Davies, G.F.; Berg, A.; Postnikoff, S.D.L.; Wilson, H.L.; Arnason, T.G.; Kusalik, A.; Harkness, T.A.A. TFPI1 Mediates Resistance to Doxorubicin in Breast Cancer Cells by Inducing a Hypoxic-Like Response. PLoS ONE 2014, 9, e84611. [Google Scholar] [CrossRef]
- Davies, G.; Lobanova, L.; Dawicki, W.; Groot, G.; Gordon, J.R.; Bowen, M.; Harkness, T.; Arnason, T. Metformin inhibits the development, and promotes the resensitization, of treatment-resistant breast cancer. PLoS ONE 2017, 12, e0187191. [Google Scholar] [CrossRef] [Green Version]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef]
- Safe, S.; Naira, V.; Karki, K. Metformin-induced anticancer activities: Recent insights. Biol. Chem. 2017, 399, 321–335. [Google Scholar] [CrossRef]
- Lu, C.-C.; Chiang, J.-H.; Tsai, F.-J.; Hsu, Y.-M.; Juan, Y.-N.; Yang, J.-S.; Chiu, H.-Y. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int. J. Oncol. 2019, 54, 1271–1281. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Yang, S.-F.; Yang, C.-K.; Tsai, H.-D.; Chen, T.-H.; Chou, M.-C.; Hsiao, Y.-H. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep. 2020, 23, 88. [Google Scholar] [CrossRef]
- Singh, A.R.; Gu, J.J.; Zhang, Q.; Torka, P.; Sundaram, S.; Mavis, C.; Hernandez-Ilizaliturri, F.J. Metformin sensitizes therapeutic agents and improves outcome in pre-clinical and clinical diffuse large B-cell lymphoma. Cancer Metab. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Barakat, H.E.; Hussein, R.R.S.; Elberry, A.A.; Zaki, M.A.; Ramadan, M.E. Factors influencing the anticancer effects of metformin on breast cancer outcomes: A systematic review and meta-analysis. Expert Rev. Anticancer Ther. 2022, 22, 415–436. [Google Scholar] [CrossRef]
- Wu, X.Y.; Xu, W.W.; Huan, X.K.; Wu, G.N.; Li, G.; Zhou, Y.H.; Najafi, M. Mechanisms of cancer cell killing by metformin: A re-view on different cell death pathways. Mol. Cell Biochem. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 2018, 17, e12723. [Google Scholar] [CrossRef]
- Justice, J.N.; Niedernhofer, L.; Robbins, P.D.; Aroda, V.R.; Espeland, M.A.; Kritchevsky, S.B.; Kuchel, G.A.; Barzilai, N. Development of Clinical Trials to Extend Healthy Lifespan. Cardiovasc. Endocrinol. Metab. 2018, 7, 80–83. [Google Scholar] [CrossRef]
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem Cells Int. 2022, 2022, 9653244. [Google Scholar] [CrossRef]
- Ito, D.; Frantz, A.M.; Modiano, J.F. Canine lymphoma as a comparative model for human non-Hodgkin lymphoma: Recent progress and applications. Vet. Immunol. Immunopathol. 2014, 159, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Seelig, D.M.; Avery, A.C.; Ehrhart, E.J.; Linden, M.A. The Comparative Diagnostic Features of Canine and Human Lympho-ma. Vet. Sci. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kritharis, A.; Beheshti, A.; Pilichowska, M.; Burgess, K.; Ricks-Santi, L.; McNiel, E.; London, C.A.; Ravi, D.; Evens, A.M. Comparative oncology DNA sequencing of canine T cell lymphoma via human hotspot panel. Oncotarget 2018, 9, 22693–22702. [Google Scholar] [CrossRef] [PubMed]
- Michel, D.; Gaunt, M.C.; Arnason, T.; El-Aneed, A. Development and validation of fast and simple flow injection analysis–tandem mass spectrometry (FIA–MS/MS) for the determination of metformin in dog serum. J. Pharm. Biomed. Anal. 2015, 107, 229–235. [Google Scholar] [CrossRef]
- Johnston, C.A.; Dickinson, V.S.M.; Alcorn, J.; Gaunt, M.C. Pharmacokinetics and oral bioavailability of metformin hydrochloride in healthy mixed-breed dogs. Am. J. Vet. Res. 2017, 78, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Aresu, L.; Ferraresso, S.; Marconato, L.; Cascione, L.; Napoli, S.; Gaudio, E.; Kwee, I.; Tarantelli, C.; Testa, A.; Maniaci, C.; et al. New molecular and therapeutic insights into canine diffuse large B-cell lymphoma elucidates the role of the dog as a model for human disease. Haematologica 2019, 104, e256–e259. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: The Reactome FI Cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar] [CrossRef]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 1–18. [Google Scholar] [CrossRef]
- Kernan, J.; Bonacci, T.; Emanuele, M.J. Who guards the guardian? Mechanisms that restrain APC/C during the cell cycle. Biochim. Biophys. Acta 2018, 1865, 1924–1933. [Google Scholar] [CrossRef]
- Zhu, G.; Spellman, P.T.; Volpe, T.; Brown, P.O.; Botstein, D.; Davis, T.N.; Futcher, B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 2000, 406, 90–94. [Google Scholar] [CrossRef]
- Haase, S.B.; Wittenberg, C. Topology and Control of the Cell-Cycle-Regulated Transcriptional Circuitry. Genetics 2014, 196, 65–90. [Google Scholar] [CrossRef]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Malo, M.E.; Postnikoff, S.D.; Arnason, T.G.; Harkness, T.A. Mitotic degradation of yeast Fkh1 by the Anaphase Promoting Complex is required for normal longevity, genomic stability and stress resistance. Aging 2016, 8, 810–830. [Google Scholar] [CrossRef]
- Sajman, J.; Zenvirth, D.; Nitzan, M.; Margalit, H.; Simpson-Lavy, K.J.; Reiss, Y.; Cohen, I.; Ravid, T.; Brandeis, M. Degradation of Ndd1 by APC/C(Cdh1) generates a feed forward loop that times mitotic protein accumulation. Nat. Commun. 2015, 6, 7075. [Google Scholar] [CrossRef]
- Kapanidou, M.; Curtis, N.L.; Bolanos-Garcia, V.M. Cdc20: At the Crossroads between Chromosome Segregation and Mitotic Exit. Trends Biochem. Sci. 2017, 42, 193–205. [Google Scholar] [CrossRef]
- VanGenderen, C.; Harkness, T.; Arnason, T.G. The role of Anaphase Promoting Complex activation, inhibition and substrates in cancer development and progression. Aging 2020, 12, 15818–15855. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharm. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef]
- Cheng, S.; Castillo, V.; Sliva, D. CDC20 associated with cancer metastasis and novel mushroom-derived CDC20 inhibitors with antimetastatic activity. Int. J. Oncol. 2019, 54, 2250–2256. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Ma, C.; Lin, H.; Tang, L.; Lian, Z.; Zhao, F.; Li, J.-M.; Zhen, B.; Pei, H.; Han, S.; et al. The anaphase promoting complex impacts repair choice by protecting ubiquitin signalling at DNA damage sites. Nat. Commun. 2017, 8, 15751. [Google Scholar] [CrossRef]
- Garzón, J.; Rodríguez, R.; Kong, Z.; Chabes, A.; Rodríguez-Acebes, S.; Méndez, J.; Moreno, S.; García-Higuera, I. Shortage of dNTPs underlies altered replication dynamics and DNA breakage in the absence of the APC/C cofactor Cdh1. Oncogene 2017, 36, 5808–5818. [Google Scholar] [CrossRef] [Green Version]
- Sansregret, L.; Patterson, J.O.; Dewhurst, S.; López-García, C.; Koch, A.; McGranahan, N.; Chao, W.C.H.; Barry, D.J.; Rowan, A.; Instrell, R.; et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017, 7, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Silvester, J.; Elliott, M.J.; Ba-Alawi, W.; Duncan, M.H.; Elia, A.C.; Mer, A.S.; Smirnov, P.; Safikhani, Z.; Haibe-Kains, B.; et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E1570–E1577. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Fu, Y.; Yu, L.; Chen, J.F.; Hansen, U.; Weng, Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004, 32, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Costa, R.H.; Lau, L.F.; Tyner, A.L.; Raychaudhuri, P. Anaphase-Promoting Complex/Cyclosome-Cdh1-Mediated Proteolysis of the Forkhead Box M1 Transcription Factor Is Critical for Regulated Entry into S Phase. Mol. Cell. Biol. 2008, 28, 5162–5171. [Google Scholar] [CrossRef]
- Kisseberth, W.C.; Nadella, M.V.P.; Breen, M.; Thomas, R.; Duke, S.E.; Murahari, S.; Kosarek, C.E.; Vernau, W.; Avery, A.C.; Burkhard, M.J.; et al. A novel canine lymphoma cell line: A translational and comparative model for lymphoma research. Leuk. Res. 2007, 31, 1709–1720. [Google Scholar] [CrossRef]
- Kastl, J.; Braun, J.; Prestel, A.; Möller, H.M.; Huhn, T.; Mayer, T. Mad2 Inhibitor-1 (M2I-1): A Small Molecule Protein–Protein Interaction Inhibitor Targeting the Mitotic Spindle Assembly Checkpoint. ACS Chem. Biol. 2015, 10, 1661–1666. [Google Scholar] [CrossRef]
- Lu, M.Z.; Li, D.Y.; Wang, X.F. Effect of metformin use on the risk and prognosis of ovarian cancer: An updated systematic review and meta-analysis. Panminerva Med. 2019. [Google Scholar] [CrossRef]
- Mekuria, A.N.; Ayele, Y.; Tola, A.; Mishore, K.M. Monotherapy with Metformin versus Sulfonylureas and Risk of Cancer in Type 2 Diabetic Patients: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2019, 2019, 7676909. [Google Scholar] [CrossRef]
- Barutello, G.; Rolih, V.; Arigoni, M.; Tarone, L.; Conti, L.; Quaglino, E.; Buracco, P.; Cavallo, F.; Riccardo, F. Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy. Int. J. Mol. Sci. 2018, 19, 799. [Google Scholar] [CrossRef]
- Abdelmegeed, S.M.; Mohammed, S. Canine mammary tumors as a model for human disease. Oncol. Lett. 2018, 15, 8195–8205. [Google Scholar] [CrossRef] [Green Version]
- Koehler, J.W.; Miller, A.D.; Miller, C.R.; Porter, B.; Aldape, K.; Beck, J.; Brat, D.; Cornax, I.; Corps, K.; Frank, C.; et al. A Revised Diagnostic Classification of Canine Glioma: Towards Validation of the Canine Glioma Patient as a Naturally Occurring Pre-clinical Model for Human Glioma. J. Neuropathol. Exp. Neurol. 2018, 77, 1039–1054. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updat. 2012, 15, 50–61. [Google Scholar] [CrossRef]
- Naylor, R.M.; van Deursen, J.M. Aneuploidy in Cancer and Aging. Annu. Rev. Genet. 2016, 50, 45–66. [Google Scholar] [CrossRef]
- Harkness, T.A.A. Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan. Int. J. Mol. Sci. 2018, 19, 1888. [Google Scholar] [CrossRef]
- Lehman, N.L.; Tibshirani, R.; Hsu, J.Y.; Natkunam, Y.; Harris, B.T.; West, R.B.; Masek, M.A.; Montgomery, K.; van de Rijn, M.; Jackson, P.K. Oncogenic regulators and substrates of the anaphase promoting complex/cyclosome are frequently overexpressed in malignant tumors. Am. J. Pathol. 2007, 170, 1793–1805. [Google Scholar] [CrossRef]
- Qiao, X.; Zhang, L.; Gamper, A.M.; Fujita, T.; Wan, Y. APC/C-Cdh1: From cell cycle to cellular differentiation and genomic integrity. Cell Cycle 2010, 9, 3904–3912. [Google Scholar] [CrossRef]
- Zhan, S.J.; Liu, B.; Linghu, H. Identifying genes as potential prognostic indicators in patients with serous ovarian cancer resistant to carboplatin using integrated bioinformatics analysis. Oncol. Rep. 2018, 39, 2653–2663. [Google Scholar] [CrossRef]
- Kelliher, C.M.; Foster, M.W.; Motta, F.C.; Deckard, A.; Soderblom, E.J.; Moseley, M.A.; Haase, S.B. Layers of regulation of cell-cycle gene expression in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2018, 29, 2644–2655. [Google Scholar] [CrossRef]
- Ostapenko, D.; Burton, J.L.; Solomon, M.J. Identification of Anaphase Promoting Complex Substrates in S. cerevisiae. PLoS ONE 2012, 7, e45895. [Google Scholar] [CrossRef]
- Ostapenko, D.; Solomon, M.J. Anaphase promoting complex–dependent degradation of transcriptional repressors Nrm1 and Yhp1 in Saccharomyces cerevisiae. Mol. Biol. Cell 2011, 22, 2175–2184. [Google Scholar] [CrossRef]
- Zhang, J.; Wan, L.; Dai, X.; Sun, Y.; Wei, W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim. Biophys. Acta 2014, 1845, 277–293. [Google Scholar] [CrossRef]
- Lu, S.; Guo, M.; Fan, Z.; Chen, Y.; Shi, X.; Gu, C.; Yang, Y. Elevated TRIP13 drives cell proliferation and drug resistance in bladder cancer. Am. J. Transl. Res. 2019, 11, 4397–4410. [Google Scholar] [PubMed]
- Wang, S.; Zhang, M.; Liang, D.; Sun, W.; Zhang, C.; Jiang, M.; Liu, J.; Li, J.; Li, C.; Yang, X.; et al. Molecular design and anti-cancer activities of small-molecule monopolar spindle 1 inhibitors: A Medicinal chemistry perspective. Eur. J. Med. Chem. 2019, 175, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Izawa, D.; Pines, J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 2014, 517, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; VanderLinden, R.; Weissmann, F.; Qiao, R.; Dube, P.; Brown, N.G.; Haselbach, D.; Zhang, W.; Sidhu, S.S.; Peters, J.-M.; et al. Cryo-EM of Mitotic Checkpoint Complex-Bound APC/C Reveals Reciprocal and Conformational Regulation of Ubiquitin Ligation. Mol. Cell 2016, 63, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Jiang, L.P.; Shen, Q.S.; Xiong, Q.X.; Zhuo, X.; Zhang, L.L.; Yu, H.J.; Guo, X.; Luo, Y.; Dong, J.; et al. NCAPH plays important roles in human colon cancer. Cell Death Dis. 2017, 8, e2680. [Google Scholar] [CrossRef]
- Hu, R.; Wang, M.-Q.; Niu, W.-B.; Wang, Y.-J.; Liu, Y.-Y.; Liu, L.-Y.; Zhong, J.; You, H.-Y.; Wu, X.-H.; Deng, N.; et al. SKA3 promotes cell proliferation and migration in cervical cancer by activating the PI3K/Akt signaling pathway. Cancer Cell Int. 2018, 18, 183. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, A.; Lin, J.; Wu, L.; Zhang, H.; Yang, X.; Wan, X.; Miao, R.; Sang, X.; Zhao, H. Mps1/TTK: A novel target and biomarker for cancer. J. Drug Target. 2017, 25, 112–118. [Google Scholar] [CrossRef]
- Zaman, G.; de Roos, J.; Libouban, M.; Prinsen, M.; de Man, J.; Buijsman, R.C.; Uitdehaag, J. TTK Inhibitors as a Targeted Therapy for CTNNB1 (β-catenin) Mutant Cancers. Mol. Cancer Ther. 2017, 16, 2609–2617. [Google Scholar] [CrossRef]
- Loddo, M.; Kingsbury, S.R.; Rashid, M.; Proctor, I.; Holt, C.; Young, J.; El-Sheikh, S.; Falzon, M.; Eward, K.L.; Prevost, T.; et al. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. Br. J. Cancer 2009, 100, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID Number | Breed | Age | B vs. T Cell Lymphoma | Stage | Chemotherapy at Time of Sampling | Clinical Response to Therapy | MET Duration | MET Dose | Time to Death After MET | Overall Survival (Diagnosis to Death) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Golden Retriever | 10 yr | B | Ⅲ | CHOP with MET | No, enlarged lymph nodes remained | 108 days | 250 mg OD | 184 days | 254 days |
| 2 | Lab Retriever mix | 5 yr | B | Ⅲ | CHOP with MET | No, enlarged lymph nodes remained | 81 days | 500 mg BID | 87 days | 277 days |
| 3 | Retriever mix | 7 yr | B | Ⅲ | DOX with MET | No, enlarged lymph nodes remained | 138 days | 500 mg BID | 143 days | 184 days |
| 4 | Lab Retriever mix | 11 yr | B | Ⅲ | CHOP failed then rescue with CCNU failed, then MET added to CHOP and remission | Yes/No, Remission attained for 8 weeks then relapse again | 84 days | 250 mg OD then 250 mg BID | 91 days | 261 days |
| Date | Canine 4 Treatment |
|---|---|
| 29 October 2014 | Initial diagnosis by referring veterinarian and initial consult with WCVM oncology. |
| 5 November 2014 | Returned to WCVM for staging, started CHOP chemotherapy protocol, received 4 treatments (1 cyce) but never achieved a complete remission. |
| 11 December 2014 | Not in remission, abandoned CHOP protocol and started rescue protocol using L-asparaginase and CCNU. Received 5 doses of CCNU along with 2 doses of L-spar. Last treatment was Marth 10, 2015. Achieved a complete remission. |
| 24 April 2015 | Out of remission. Fine needle aspirate of lymph node confirmed lymphoma in nodes. |
| 28 April 2015 | Enrolled in metformin study, full bloodwork (CBC/biochemistry/urinalysis), abdominal ultrasound, chest X-ray, lymph node removal and skin biopsy. |
| 29 April 2015 | Started CHOP again-received first dose of Vincristine and metformin. Went through 2 cycles of CHOP (8 treatments total). Achieved strong partial remission but lymph nodes never completely returned to normal, so there were opportunities to take samples. |
| 13 July 2015 | No longer responding to CHOP protocol, progressive disease in lymph nodes, received one dose of CCNU. |
| 27 July 2015 | Euthanasia. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnason, T.G.; MacDonald-Dickinson, V.; Gaunt, M.C.; Davies, G.F.; Lobanova, L.; Trost, B.; Gillespie, Z.E.; Waldner, M.; Baldwin, P.; Borrowman, D.; et al. Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma. Cancers 2022, 14, 4215. https://doi.org/10.3390/cancers14174215
Arnason TG, MacDonald-Dickinson V, Gaunt MC, Davies GF, Lobanova L, Trost B, Gillespie ZE, Waldner M, Baldwin P, Borrowman D, et al. Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma. Cancers. 2022; 14(17):4215. https://doi.org/10.3390/cancers14174215
Chicago/Turabian StyleArnason, Terra G., Valerie MacDonald-Dickinson, Matthew Casey Gaunt, Gerald F. Davies, Liubov Lobanova, Brett Trost, Zoe E. Gillespie, Matthew Waldner, Paige Baldwin, Devon Borrowman, and et al. 2022. "Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma" Cancers 14, no. 17: 4215. https://doi.org/10.3390/cancers14174215
APA StyleArnason, T. G., MacDonald-Dickinson, V., Gaunt, M. C., Davies, G. F., Lobanova, L., Trost, B., Gillespie, Z. E., Waldner, M., Baldwin, P., Borrowman, D., Marwood, H., Vizeacoumar, F. S., Vizeacoumar, F. J., Eskiw, C. H., Kusalik, A., & Harkness, T. A. A. (2022). Activation of the Anaphase Promoting Complex Reverses Multiple Drug Resistant Cancer in a Canine Model of Multiple Drug Resistant Lymphoma. Cancers, 14(17), 4215. https://doi.org/10.3390/cancers14174215