
Challenges for Triple Negative Breast Cancer Treatment: Defeating Heterogeneity and Cancer Stemness


Abstract
:Simple Summary
Abstract
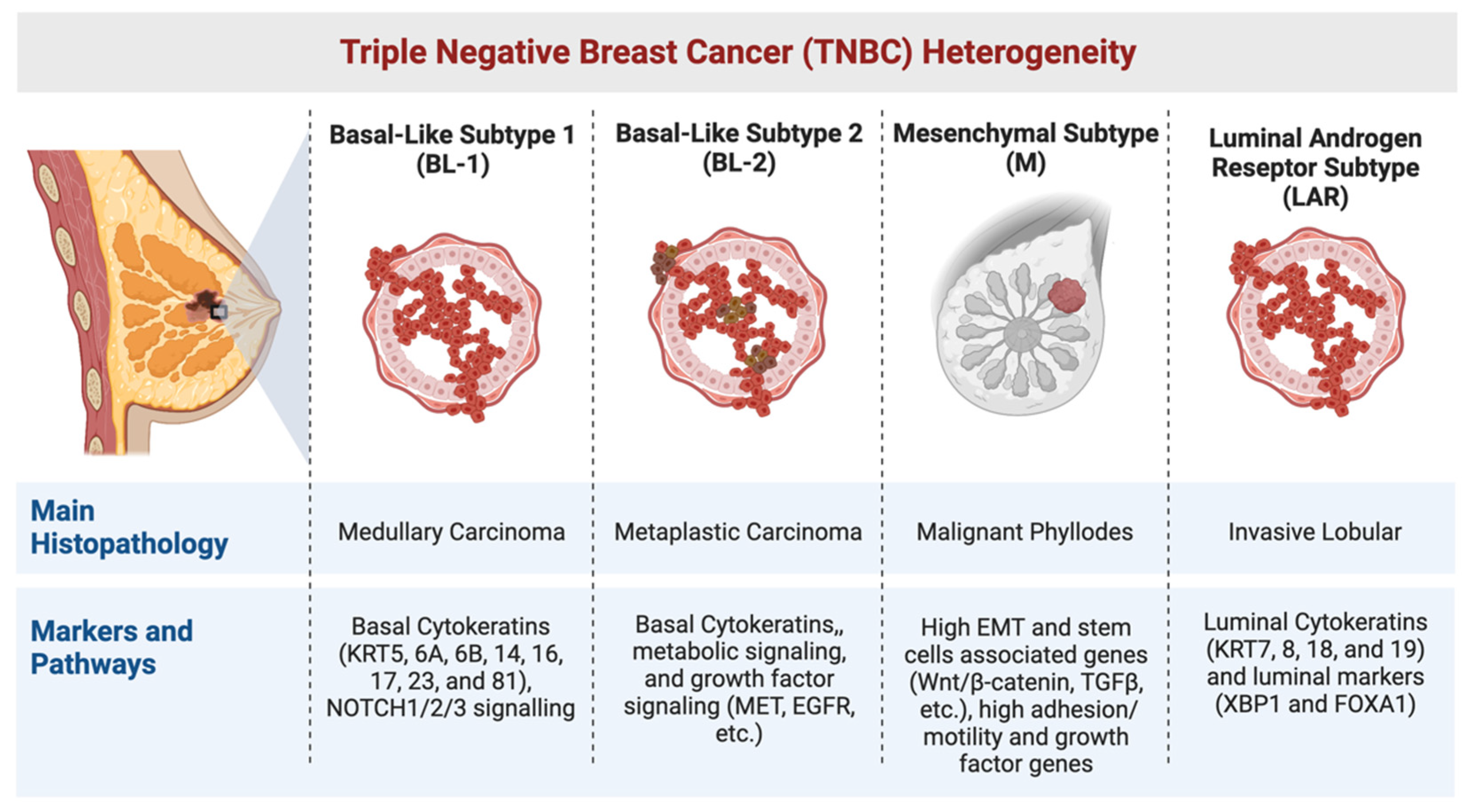
1. Heterogeneity in TNBC
1.1. The Basal-like (BL) Subtypes
1.2. The Mesenchymal (M) Subtype
1.3. The Luminal Androgen Receptor (LAR) Subtype
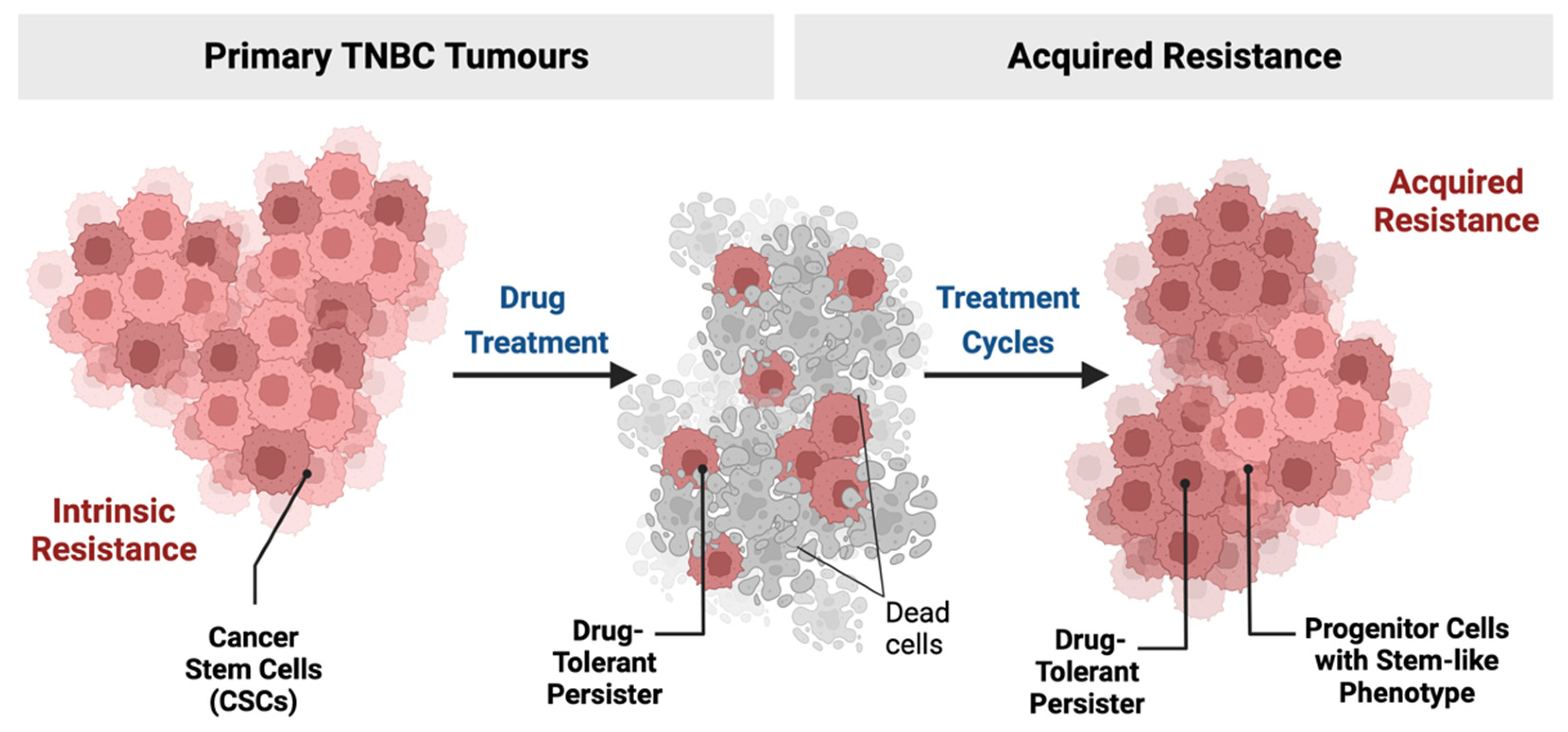
2. TNBC and Drug-Resistant Cells
2.1. Stem Cells and Cellular Origin of TNBC
2.2. Persister Cells in TNBC
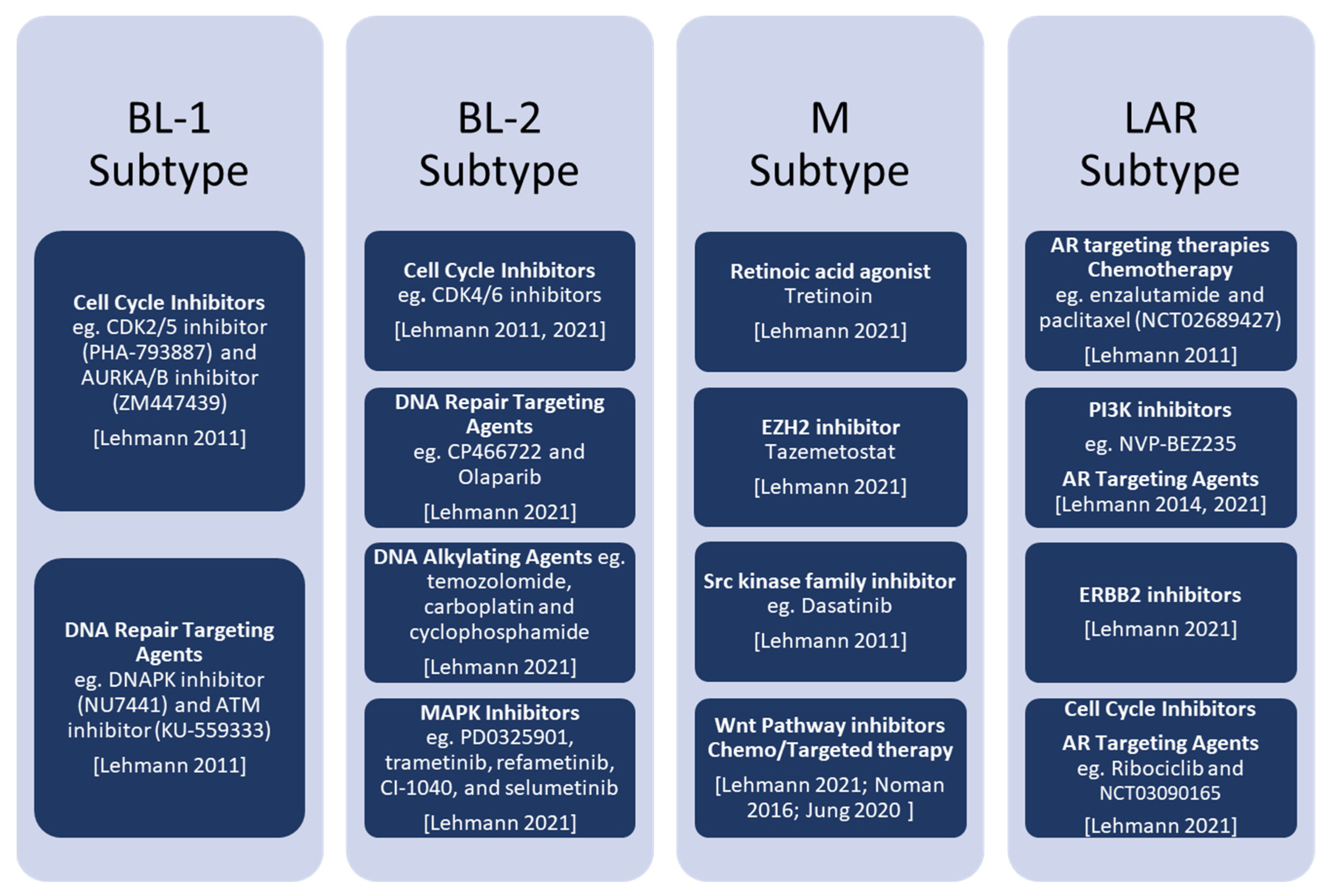
3. Novel Therapeutic Approaches for TNBC
3.1. BL Subtypes
3.2. M Subtype
3.3. LAR Subtype
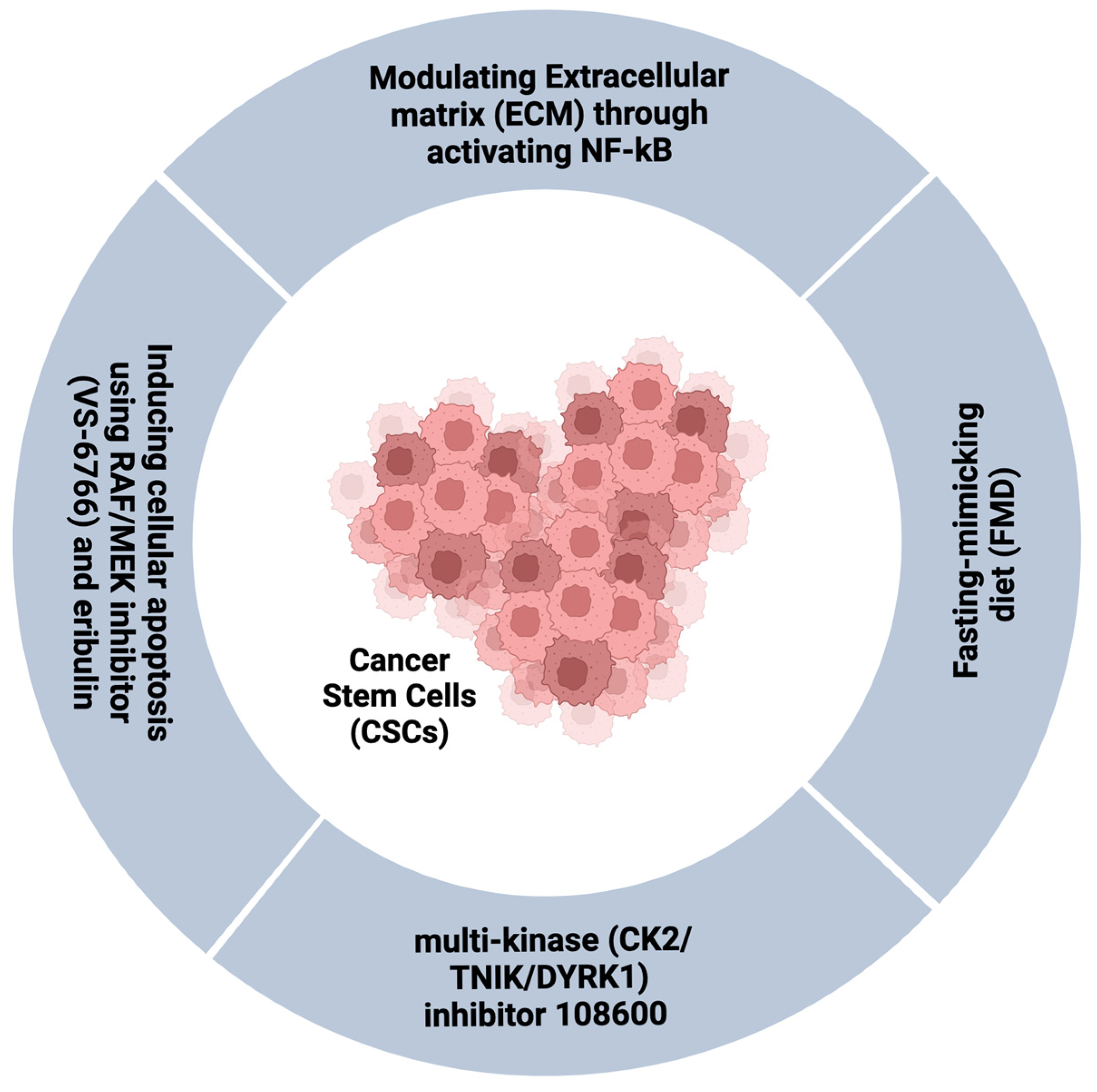
3.4. Targeting the CSCs and/or Persistent Cell Population in TNBC
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rakha, E.A.; Elsheikh, S.E.; Aleskandarany, M.A.; Habashi, H.O.; Green, A.R.; Powe, D.G.; El-Sayed, M.E.; Benhasouna, A.; Brunet, J.S.; LAkslen, A.; et al. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Clin. Cancer Res. 2009, 15, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the treatment of women with early breast cancer: Highlights of the st gallen international expert consensus on the primary therapy of early breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Colleoni, M.; Rotmensz, N.; Robertson, C.; Orlando, L.; Viale, G.; Renne, G.; Luini, A.; Veronesi, P.; Intra, M.; Orecchia, R.; et al. Very young women (<35 years) with operable breast cancer: Features of disease at presentation. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2002, 13, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.L.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the national cancer institute’s surveillance, epidemiology, and end results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Reddy, S.M.; Barcenas, C.H.; Sinha, A.K.; Hsu, L.; Moulder, S.L.; Tripathy, D.; Hortobagyi, G.N.; Valero, V. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease free at 5 years and relationship with low hormone receptor positivity. Br. J. Cancer 2017, 118, 17–23. [Google Scholar] [CrossRef]
- Liu, Y.; Xin, T.; Huang, D.-Y.; Shen, W.-X.; Li, L.; Lv, Y.-J.; Jin, Y.-H.; Song, X.-W.; Teng, C.; Jiang, Q.-Y. Prognosis in very young women with triple-negative breast cancer: Retrospective study of 216 cases. Med. Oncol. 2014, 31, 222. [Google Scholar] [CrossRef]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Rakha, E.A.; Tan, D.S.P.; Foulkes, W.D.; Ellis, I.O.; Tutt, A.; Nielsen, T.O.; Reis-Filho, J.S. Are triple-negative tumours and basal-like breast cancer synonymous? Breast Cancer Res. 2007, 9, 404. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer: Who Should Receive Neoadjuvant Chemotherapy? Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2021, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Jiang, Y.-Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.-D.; Liu, Y.-R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Z.; Qu, X.; Zhang, Z. Prognostic value of tumor-infiltrating lymphocytes in patients with triple-negative breast cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 179. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Kochi, M.; Iwamoto, T.; Niikura, N.; Bianchini, G.; Masuda, S.; Mizoo, T.; Nogami, T.; Shien, T.; Motoki, T.; Taira, N.; et al. Tumour-infiltrating lymphocytes (TILs)-related genomic signature predicts chemotherapy response in breast cancer. Breast Cancer Res. Treat. 2018, 167, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef]
- Koleckova, M.; Vomackova, K.; Kolar, Z. Molecular Prognostic and Predictive Markers in Triple—Negative Breast Cancer. In Breast Cancer—Evolving Challenges and Next Frontiers; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Abd El-Rehim, D.M.; Pinder, S.E.; Paish, C.E.; Bell, J.; Blamey, R.W.; Robertson, J.F.R.; Nicholson, R.I.; Ellis, I.O. Expression of luminal and basal cytokeratins in human breast carcinoma. J. Pathol. 2004, 203, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.A.; Kung, Y.; Salari, K.; Holcomb, I.N.; Pollack, J.R. Genomic instability in breast cancer: Pathogenesis and clinical implications. Mol. Oncol. 2010, 4, 255–266. [Google Scholar] [CrossRef]
- Bauer, J.A.; Chakravarthy, A.B.; Rosenbluth, J.M.; Mi, D.; Seeley, E.H.; Granja-Ingram, N.D.M.; Olivares, M.G.; Kelley, M.C.; Mayer, I.A.; Meszoely, I.M.; et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin. Cancer Res. 2010, 16, 681–690. [Google Scholar] [CrossRef]
- Juul, N.; Szallasi, Z.; Eklund, A.C.; Li, Q.; Burrell, R.A.; Gerlinger, M.; Valero, V.; Andreopoulou, E.; Esteva, F.J.; Symmans, W.F.; et al. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: A retrospective analysis of five clinical trials. Lancet Oncol. 2010, 11, 358–365. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Paroni, G.; Zanetti, A.; Barzago, M.M.; Kurosaki, M.; Guarrera, L.; Fratelli, M.; Troiani, M.; Ubezio, P.; Bolis, M.; Vallerga, A.; et al. Retinoic Acid Sensitivity of Triple-Negative Breast Cancer Cells Characterized by Constitutive Activation of the notch1 Pathway: The Role of Rarβ. Cancers 2020, 12, 3027. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST Domain Mutations in Notch Receptors Comprise an Oncogenic Driver Segment in Triple-Negative Breast Cancer Sensitive to a γ-Secretase Inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Q.; Zou, Y.; Chen, H.; Qi, L.; Chen, Y. Stem cells and cellular origins of breast cancer: Updates in the rationale, controversies, and therapeutic implications. Front. Oncol. 2019, 9, 820. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Clevers, H. Cancer therapy: Defining stemness. Nature 2016, 534, 176–177. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54–86. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef]
- Dey, D.; Saxena, M.; Paranjape, A.N.; Krishnan, V.; Giraddi, R.; Kumar, M.V.; Mukherjee, G.; Rangarajan, A. Phenotypic and Functional Characterization of Human Mammary Stem/Progenitor Cells in Long Term Culture. PLoS ONE 2009, 4, e5329. [Google Scholar] [CrossRef] [PubMed]
- Uchôa, D.D.M.; Graudenz, M.S.; Callegari-Jacques, S.M.; Hartmann, C.R.; Ferreira, B.P.; Fitarelli-Kiehl, M.; Edelweiss, M.I. Expression of cancer stem cell markers in basal and penta-negative breast carcinomas—A study of a series of triple-negative tumors. Pathol. Res. Pract. 2014, 210, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Perrone, G.; Gaeta, L.M.; Zagami, M.; Nasorri, F.; Coppola, R.; Borzomati, D.; Bartolozzi, F.; Altomare, V.; Trodella, L.; Tonini, G.; et al. In Situ Identification of CD44+/CD24− Cancer Cells in Primary Human Breast Carcinomas. PLoS ONE 2012, 7, 43110. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Vaillant, F.; Simpson, K.; Stingl, J.; Smyth, G.; Asselin-Labat, M.-L.; Wu, L.; Lindeman, G.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2013, 2, 78–91. [Google Scholar] [CrossRef]
- Honeth, G.; Bendahl, P.-O.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, A.; Hegardt, C. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef]
- Hua, Z.; White, J.; Zhou, J. Cancer stem cells in TNBC. Semin. Cancer Biol. 2022, 82, 26–34. [Google Scholar] [CrossRef]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44+/CD24− population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef]
- Shadbad, M.A.; Hosseinkhani, N.; Asadzadeh, Z.; Derakhshani, A.; Ahangar, N.K.; Hemmat, N.; Lotfinejad, P.; Brunetti, O.; Silvestris, N.; Baradaran, B. A Systematic Review to Clarify the Prognostic Values of CD44 and CD44+CD24- Phenotype in Triple-Negative Breast Cancer Patients: Lessons Learned and the Road Ahead. Front. Oncol. 2021, 11, 3096. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Song, Y.; Wang, S.; Huang, X.; Xuan, Q.; Kang, X.; Zhang, Q. CD44+/CD24-phenotype predicts a poor prognosis in triple-negative breast cancer. Oncol. Lett. 2017, 14, 5890–5898. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.-F.; Heymann, D. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Osisami, M.; Keller, E.T. Mechanisms of Metastatic Tumor Dormancy. J. Clin. Med. 2013, 2, 136. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- de Angelis, M.L.; Francescangeli, F.; la Torre, F.; Zeuner, A. Stem cell plasticity and dormancy in the development of cancer therapy resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.; Francescangeli, F.; Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers 2019, 11, 1569. [Google Scholar] [CrossRef] [PubMed]
- Gomez, K.; Rabadan, R. A persistent look at how tumours evade therapy. Nature 2021, 596, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Wu, X.; Yang, B.; Yang, X.; Zhang, D.; Qing, G. Stem cell characteristics of dormant cells and cisplatin-induced effects on the stemness of epithelial ovarian cancer cells. Mol. Med. Rep. 2014, 10, 2495–2504. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef]
- de Oliveira, W.A.A.; Moens, S.; El Laithy, Y.; van der Veer, B.K.; Athanasouli, P.; Cortesi, E.E.; Baietti, M.F.; Koh, K.P.; Ventura, J.-J.; Amant, F.; et al. Wnt/β-Catenin Inhibition Disrupts Carboplatin Resistance in Isogenic Models of Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 2754. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Qayoom, H.; Wani, N.A.; Alshehri, B.; Mir, M.A. An insight into the cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol. 2021, 17, 4185–4206. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.S.; Uddin, M.T.; Rahman, M.Z.; Nayeem, M.J.; Alam, S.S.; Khatun, Z.; Wahiduzzaman, M.; Sultana, A.; Ali, M.Y.; Barua, D.; et al. Overexpression of sonic hedgehog in the triple negative breast cancer: Clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci. Rep. 2016, 6, 18830. [Google Scholar] [CrossRef]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef]
- Han, J.; Yun, J.; Quan, M.; Kang, W.; Jung, J.-G.; Heo, W.; Li, S.; Lee, K.J.; Son, H.-Y.; Kim, J.H.; et al. JAK2 regulates paclitaxel resistance in triple negative breast cancers. J. Mol. Med. 2021, 99, 1783–1795. [Google Scholar] [CrossRef]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Wu, L.; Yang, X. Targeting the Hippo Pathway for Breast Cancer Therapy. Cancers 2018, 10, 422. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Chakraborty, S.; Bhattacharya, R.; Chowdhury, G. Senescence-Induced Chemoresistance in Triple Negative Breast Cancer and Evolution-Based Treatment Strategies. Front. Oncol. 2021, 11, 674354. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Berger, S.L. Senescence Elicits Stemness: A Surprising Mechanism for Cancer Relapse. Cell Metab. 2018, 27, 710–711. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348. [Google Scholar] [CrossRef]
- Sulaiman, A.; McGarry, S.; Chilumula, S.C.; Kandunuri, R.; Vinod, V. Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC. Biomedicines 2021, 9, 1386. [Google Scholar] [CrossRef]
- Xia, L.Y.; Hu, Q.L.; Zhang, J.; Xu, W.Y.; Li, X.S. Survival outcomes of neoadjuvant versus adjuvant chemotherapy in triple-negative breast cancer: A meta-analysis of 36,480 cases. World J. Surg. Oncol. 2020, 18, 129. [Google Scholar] [CrossRef]
- King, T.A.; Morrow, M. Surgical issues in patients with breast cancer receiving neoadjuvant chemotherapy. Nat. Rev. Clin. Oncol. 2015, 12, 335–343. [Google Scholar] [CrossRef]
- Schmidt, M.; Heimes, A.-S. Immunomodulating Therapies in Breast Cancer—From Prognosis to Clinical Practice. Cancers 2021, 13, 4883. [Google Scholar] [CrossRef]
- Zacharakis, N.; Huq, L.M.; Seitter, S.J.; Kim, S.P.; Gartner, J.J.; Sindiri, S.; Hill, V.K.; Li, Y.F.; Paria, B.C.; Ray, S.; et al. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol. 2022, 40, 1741–1754. [Google Scholar] [CrossRef]
- Wu, Q.; Nie, D.Y.; Ba-Alawi, W.; Ji, Y.; Zhang, Z.; Cruickshank, J.; Haight, J.; Ciamponi, F.E.; Chen, J.; Duan, S.; et al. PRMT inhibition induces a viral mimicry response in triple-negative breast cancer. Nat. Chem. Biol. 2022, 18, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.-M.; Landragin, C.; Trouchet, A.; Bento, S.T.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 conditions chemotolerance in triple-negative breast cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ribociclib and Bicalutamide in AR+ TNBC—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03090165 (accessed on 3 January 2022).
- Hennessy, B.T.; Gonzalez-Angulo, A.-M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.-S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2009, 223, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Thomas, D.; Emmons, A.; Giordano, T.J.; Kleer, C.G. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin. Cancer Res. 2008, 14, 4038–4044. [Google Scholar] [CrossRef]
- Koval, A.; Katanaev, V.L. Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 2018, 8, 7329. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR+ Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef]
- Salvadori, G.; Zanardi, F.; Iannelli, F.; Lobefaro, R.; Vernieri, C.; Longo, V.D. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 2021, 33, 2247. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, M.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.-K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef] [PubMed]
- Drain, A.P.; Zahir, N.; Northey, J.J.; Zhang, H.; Huang, P.-J.; Maller, O.; Lakins, J.N.; Yu, X.; Leight, J.L.; Alston-Mills, B.P.; et al. Matrix compliance permits NF-κB activation to drive therapy resistance in breast cancer. J. Exp. Med. 2021, 218, e20191360. [Google Scholar] [CrossRef]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.R.C.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 inhibition by 108600 suppresses triple negative breast cancer stem cells and chemotherapy-resistant disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Horinaka, M.; Sukeno, M.; Morita, M.; Yasuda, S.; Nishimoto, E.; Konishi, E.; Sakai, T. Novel RAF/MEK inhibitor CH5126766/VS-6766 has efficacy in combination with eribulin for the treatment of triple-negative breast cancer. Cancer Sci. 2021, 112, 4166–4175. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, R.; Ordóñez-Morán, P.; Allegrucci, C. Challenges for Triple Negative Breast Cancer Treatment: Defeating Heterogeneity and Cancer Stemness. Cancers 2022, 14, 4280. https://doi.org/10.3390/cancers14174280
Mahmoud R, Ordóñez-Morán P, Allegrucci C. Challenges for Triple Negative Breast Cancer Treatment: Defeating Heterogeneity and Cancer Stemness. Cancers. 2022; 14(17):4280. https://doi.org/10.3390/cancers14174280
Chicago/Turabian StyleMahmoud, Rinad, Paloma Ordóñez-Morán, and Cinzia Allegrucci. 2022. "Challenges for Triple Negative Breast Cancer Treatment: Defeating Heterogeneity and Cancer Stemness" Cancers 14, no. 17: 4280. https://doi.org/10.3390/cancers14174280
APA StyleMahmoud, R., Ordóñez-Morán, P., & Allegrucci, C. (2022). Challenges for Triple Negative Breast Cancer Treatment: Defeating Heterogeneity and Cancer Stemness. Cancers, 14(17), 4280. https://doi.org/10.3390/cancers14174280