
Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention

 , , , ,
, , , , {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. TNBC—The Triple Threat
3. AR Signaling/Pathway
4. AR Pathway as a Therapeutic Target for TNBC
5. A Double-Edged Sword: Controversial Role of ARs in ER+ Breast Cancer and TNBC
6. QNBC—The Quadruple Threat
7. A New Racial Disparity in Breast Cancer: Characterization of QNBC Disparity in Women of African versus European Ancestry
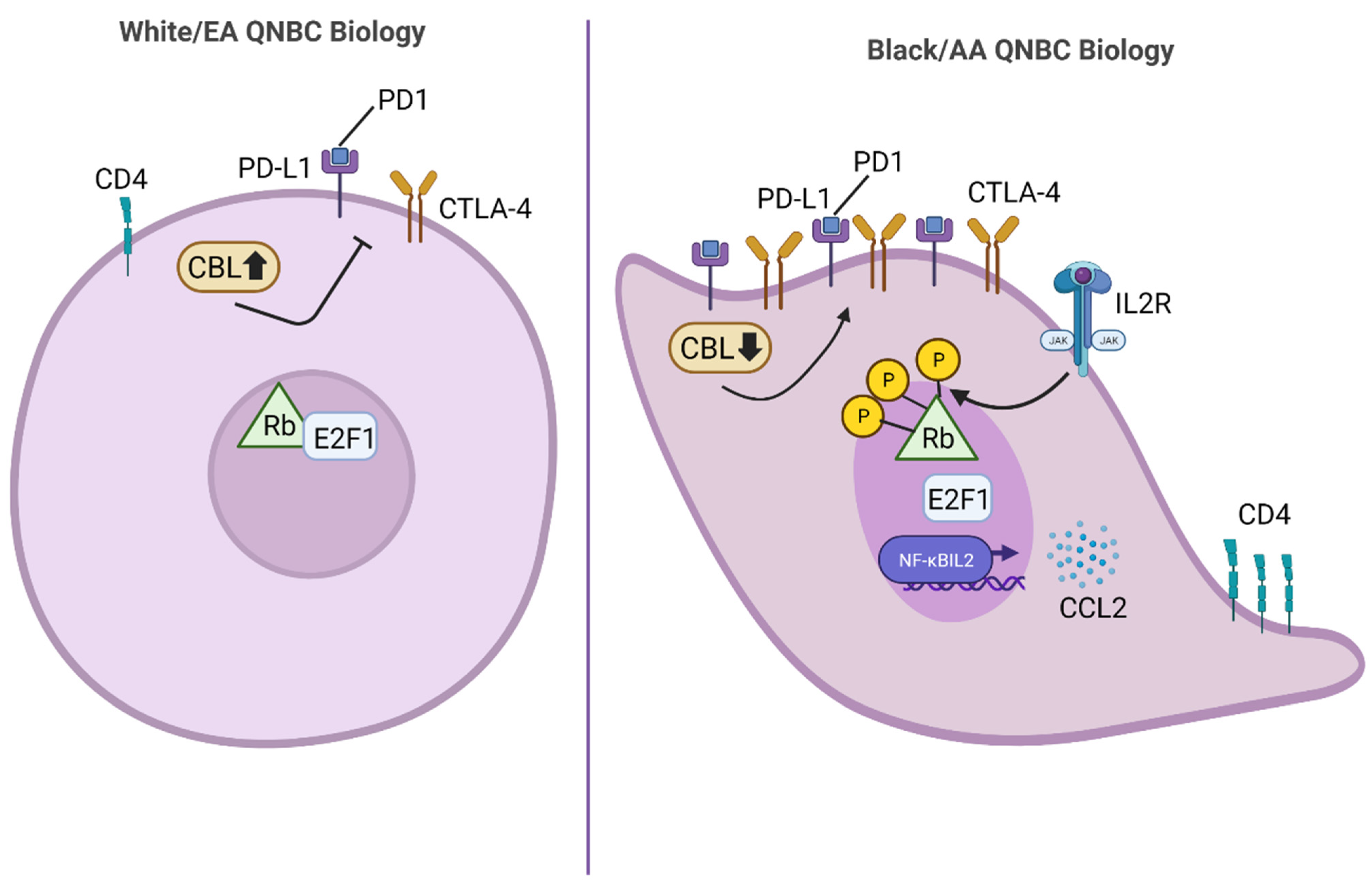
8. Black/AA versus White/EA QNBC Biology: Novel Therapeutic Strategies to Reduce the Racially Disparate Burden in QNBC
9. Spotlight on KIFC1: A Promising Target for QNBC in Women of West African Descent
10. Black/AA versus White/EA QNBC Epigenomics: The Potential Role of Epigenetic Modifications in the Racially Disparate Burden in QNBC
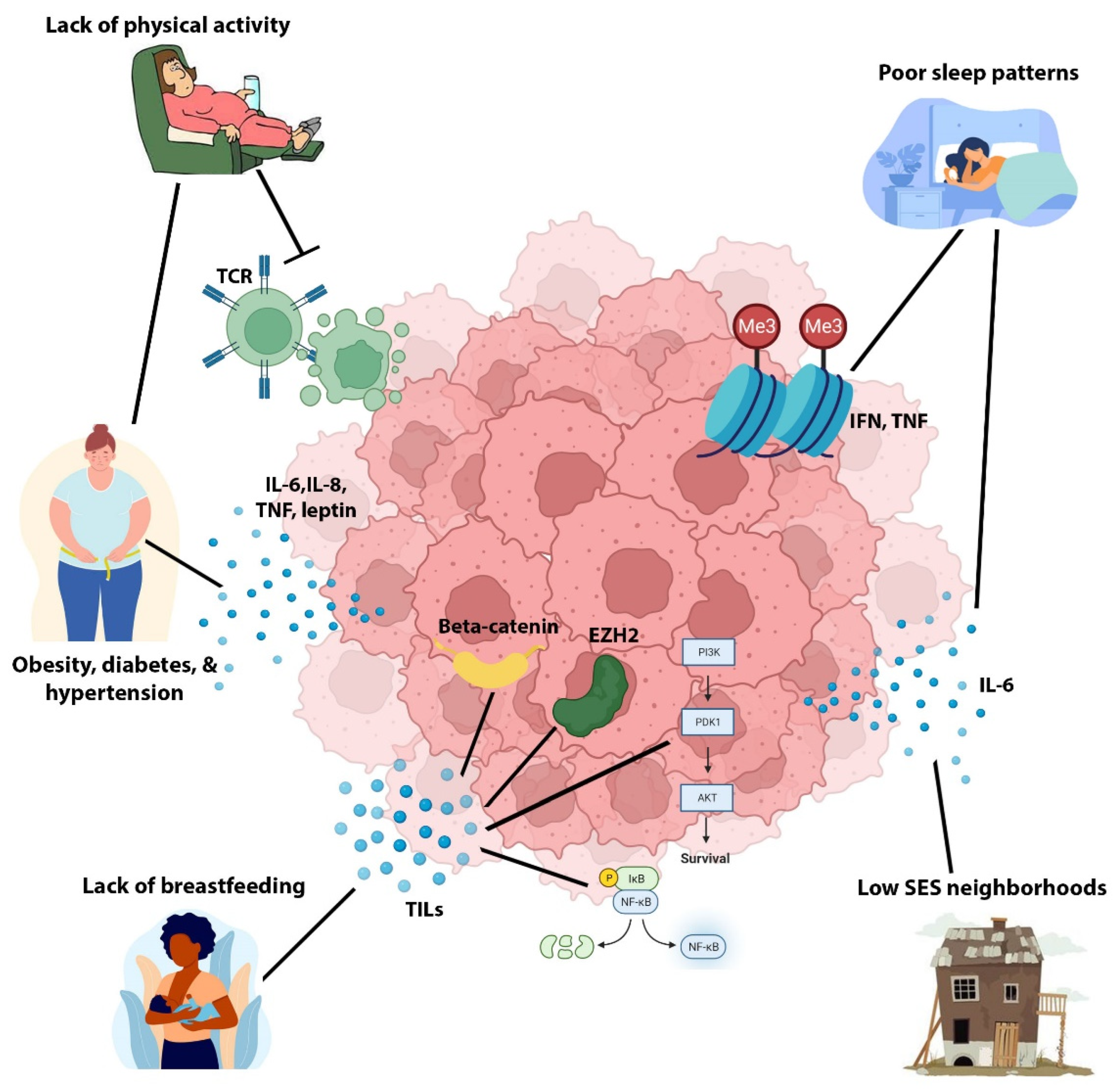
11. Black/AA versus White/EA QNBC Non-Biology: The Potential Role of Non-Genetic Risk Factors in the Racially Disparate Burden in QNBC
12. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, E.; Ulaner, G.A.; Chin, S.F. Molecular Classification of Breast Cancer. PET Clin. 2018, 13, 325–338. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Copley, B.; Niu, Q.; Liu, F.; Johnson, J.A.; Sutton, T.; Khramtsova, G.; Sveen, E.; Yoshimatsu, T.F.; Zheng, Y.; et al. Racial disparities in survival outcomes among breast cancer patients by molecular subtypes. Breast Cancer Res. Treat. 2021, 185, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Troester, M.A.; Sun, X.; Allott, E.H.; Geradts, J.; Cohen, S.M.; Tse, C.K.; Kirk, E.L.; Thorne, L.B.; Mathews, M.; Li, Y.; et al. Racial Differences in PAM50 Subtypes in the Carolina Breast Cancer Study. J. Natl. Cancer Inst. 2018, 110, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef]
- Lund, M.J.; Trivers, K.F.; Porter, P.L.; Coates, R.J.; Leyland-Jones, B.; Brawley, O.W.; Flagg, E.W.; O’Regan, R.M.; Gabram, S.G.; Eley, J.W. Race and triple negative threats to breast cancer survival: A population-based study in Atlanta, GA. Breast Cancer Res. Treat. 2009, 113, 357–370. [Google Scholar] [CrossRef]
- Bhattarai, S.; Klimov, S.; Mittal, K.; Krishnamurti, U.; Li, X.B.; Oprea-Ilies, G.; Wetherilt, C.S.; Riaz, A.; Aleskandarany, M.A.; Green, A.R.; et al. Prognostic Role of Androgen Receptor in Triple Negative Breast Cancer: A Multi-Institutional Study. Cancers 2019, 11, 995. [Google Scholar] [CrossRef]
- Davis, M.; Tripathi, S.; Hughley, R.; He, Q.; Bae, S.; Karanam, B.; Martini, R.; Newman, L.; Colomb, W.; Grizzle, W.; et al. AR negative triple negative or “quadruple negative” breast cancers in African American women have an enriched basal and immune signature. PLoS ONE 2018, 13, e0196909. [Google Scholar] [CrossRef] [Green Version]
- Newman, L.A.; Jenkins, B.; Chen, Y.; Oppong, J.K.; Adjei, E.; Jibril, A.S.; Hoda, S.; Cheng, E.; Chitale, D.; Bensenhaver, J.M.; et al. Hereditary Susceptibility for Triple Negative Breast Cancer Associated With Western Sub-Saharan African Ancestry: Results From an International Surgical Breast Cancer Collaborative. Ann. Surg. 2019, 270, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Fassan, M.; Cascione, L.; Guler, G.; Balci, S.; Irkkan, C.; Paisie, C.; Lovat, F.; Morrison, C.; Zhang, J.; et al. Androgen receptor status is a prognostic marker in non-basal triple negative breast cancers and determines novel therapeutic options. PLoS ONE 2014, 9, e88525. [Google Scholar] [CrossRef] [PubMed]
- Hon, J.D.; Singh, B.; Sahin, A.; Du, G.; Wang, J.; Wang, V.Y.; Deng, F.M.; Zhang, D.Y.; Monaco, M.E.; Lee, P. Breast cancer molecular subtypes: From TNBC to QNBC. Am. J. Cancer Res. 2016, 6, 1864–1872. [Google Scholar]
- Huang, M.; Wu, J.; Ling, R.; Li, N. Quadruple negative breast cancer. Breast Cancer 2020, 27, 527–533. [Google Scholar] [CrossRef]
- Angajala, A.; Mothershed, E.; Davis, M.B.; Tripathi, S.; He, Q.; Bedi, D.; Dean-Colomb, W.; Yates, C. Quadruple Negative Breast Cancers (QNBC) Demonstrate Subtype Consistency among Primary and Recurrent or Metastatic Breast Cancer. Transl. Oncol. 2019, 12, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Bonotto, M.; Gerratana, L.; Poletto, E.; Driol, P.; Giangreco, M.; Russo, S.; Minisini, A.M.; Andreetta, C.; Mansutti, M.; Pisa, F.E.; et al. Measures of outcome in metastatic breast cancer: Insights from a real-world scenario. Oncologist 2014, 19, 608–615. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.; Rida, P.C.G.; Aneja, R. Tackling intra- and inter-tumor heterogeneity to combat triple negative breast cancer. Front. Biosci. (Landmark Ed.) 2017, 22, 1549–1580. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef]
- Keenan, T.; Moy, B.; Mroz, E.A.; Ross, K.; Niemierko, A.; Rocco, J.W.; Isakoff, S.; Ellisen, L.W.; Bardia, A. Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. J. Clin. Oncol. 2015, 33, 3621–3627. [Google Scholar] [CrossRef]
- Anestis, A.; Zoi, I.; Papavassiliou, A.G.; Karamouzis, M.V. Androgen Receptor in Breast Cancer-Clinical and Preclinical Research Insights. Molecules 2020, 25, 358. [Google Scholar] [CrossRef]
- Zhou, X. Roles of androgen receptor in male and female reproduction: Lessons from global and cell-specific androgen receptor knockout (ARKO) mice. J. Androl. 2010, 31, 235–243. [Google Scholar] [CrossRef]
- Walters, K.A.; Simanainen, U.; Handelsman, D.J. Molecular insights into androgen actions in male and female reproductive function from androgen receptor knockout models. Hum. Reprod. Update 2010, 16, 543–558. [Google Scholar] [CrossRef]
- Zhou, J.; Ng, S.; Adesanya-Famuiya, O.; Anderson, K.; Bondy, C.A. Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression. FASEB J. 2000, 14, 1725–1730. [Google Scholar] [CrossRef]
- Dimitrakakis, C.; Zhou, J.; Wang, J.; Belanger, A.; LaBrie, F.; Cheng, C.; Powell, D.; Bondy, C. A physiologic role for testosterone in limiting estrogenic stimulation of the breast. Menopause 2003, 10, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, K.; Daxenbichler, G. The Role of Androgens in Normal and Malignant Breast Tissue. Breast Care 2008, 3, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Venema, C.M.; Bense, R.D.; Steenbruggen, T.G.; Nienhuis, H.H.; Qiu, S.Q.; van Kruchten, M.; Brown, M.; Tamimi, R.M.; Hospers, G.A.P.; Schroder, C.P.; et al. Consideration of breast cancer subtype in targeting the androgen receptor. Pharmacol. Ther. 2019, 200, 135–147. [Google Scholar] [CrossRef]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Alahari, S.K. Combination treatment of bicalutamide and curcumin has a strong therapeutic effect on androgen receptor-positive triple-negative breast cancers. Anticancer Drugs 2020, 31, 359–367. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Lind, H.T.; Spoelstra, N.S.; Babbs, B.L.; Heinz, R.E.; Elias, A.; Jedlicka, P.; Jacobsen, B.M.; et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol. Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef]
- Caiazza, F.; Murray, A.; Madden, S.F.; Synnott, N.C.; Ryan, E.J.; O’Donovan, N.; Crown, J.; Duffy, M.J. Preclinical evaluation of the AR inhibitor enzalutamide in triple-negative breast cancer cells. Endocr. Relat. Cancer 2016, 23, 323–334. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Albiges, L.; Loriot, Y.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer. Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef]
- Jinna, N.; Rida, P.; Smart, M.; LaBarge, M.; Jovanovic-Talisman, T.; Natarajan, R.; Seewaldt, V. Adaptation to Hypoxia May Promote Therapeutic Resistance to Androgen Receptor Inhibition in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 8844. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, A.R.; Chandler, B.; Olsen, E.; Wilder-Romans, K.; Moubadder, L.; Liu, M.; Pesch, A.M.; Zhang, A.; Ritter, C.; Ward, S.T.; et al. Seviteronel, a Novel CYP17 Lyase Inhibitor and Androgen Receptor Antagonist, Radiosensitizes AR-Positive Triple Negative Breast Cancer Cells. Front. Endocrinol. 2020, 11, 35. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef]
- Aleskandarany, M.A.; Rakha, E.A.; Ahmed, M.A.; Powe, D.G.; Ellis, I.O.; Green, A.R. Clinicopathologic and molecular significance of phospho-Akt expression in early invasive breast cancer. Breast Cancer Res. Treat. 2011, 127, 407–416. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR(+) Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef]
- Birrell, S.N.; Butler, L.M.; Harris, J.M.; Buchanan, G.; Tilley, W.D. Disruption of androgen receptor signaling by synthetic progestins may increase risk of developing breast cancer. FASEB J. 2007, 21, 2285–2293. [Google Scholar] [CrossRef]
- Zhu, X.; Li, H.; Liu, J.P.; Funder, J.W. Androgen stimulates mitogen-activated protein kinase in human breast cancer cells. Mol. Cell. Endocrinol. 1999, 152, 199–206. [Google Scholar] [CrossRef]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Xie, B. The role of androgens in mammary carcinogenesis. Ital. J. Anat. Embryol. 2001, 106, 111–125. [Google Scholar] [PubMed]
- Hickey, T.; Robinson, J.; Carroll, J.; Tilley, W. Minireview: The androgen receptor in breast tissues: Growth inhibitor, tumor suppressor, oncogene? Mol. Endocrinol. 2012, 26, 1252–1267. [Google Scholar] [CrossRef]
- Wilson, J.D.; Griffin, J.E.; Leshin, M.; George, F.W. Role of gonadal hormones in development of the sexual phenotypes. Hum. Genet. 1981, 58, 78–84. [Google Scholar] [CrossRef]
- Yu, K.-D.; Zhu, R.; Zhan, M.; Rodriguez, A.A.; Yang, W.; Wong, S.; Makris, A.; Lehmann, B.D.; Chen, X.; Mayer, I. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 2013, 19, 2723–2733. [Google Scholar] [CrossRef]
- Birrell, S.; Hall, R.; Tilley, W. Role of the androgen receptor in human breast cancer. J. Mammary Gland. Biol. Neoplasia 1998, 3, 95–103. [Google Scholar] [CrossRef]
- Garay, J.P.; Karakas, B.; Abukhdeir, A.M.; Cosgrove, D.P.; Gustin, J.P.; Higgins, M.J.; Konishi, H.; Konishi, Y.; Lauring, J.; Mohseni, M. The growth response to androgen receptor signaling in ER α-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012, 14, R27. [Google Scholar] [CrossRef]
- Kuenen-Boumeester, V.; Van der Kwast, T.H.; Claassen, C.C.; Look, M.P.; Liem, G.S.; Klijn, J.G.; Henzen-Logmans, S.C. The clinical significance of androgen receptors in breast cancer and their relation to histological and cell biological parameters. Eur. J. Cancer 1996, 32, 1560–1565. [Google Scholar] [CrossRef]
- Ogawa, Y.; Hai, E.; Matsumoto, K.; Ikeda, K.; Tokunaga, S.; Nagahara, H.; Sakurai, K.; Inoue, T.; Nishiguchi, Y. Androgen receptor expression in breast cancer: Relationship with clinicopathological factors and biomarkers. Int. J. Clin. Oncol. 2008, 13, 431–435. [Google Scholar] [CrossRef]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.H.; Choi, S.Y.; Lee, J.H.; Park, B.W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Mirzania, M. Approach to the Triple Negative Breast Cancer in new drugs area. Int. J. Hematol.-Oncol. Stem Cell Res. 2016, 10, 115. [Google Scholar] [PubMed]
- Sutton, L.M.; Cao, D.; Sarode, V.; Molberg, K.H.; Torgbe, K.; Haley, B.; Peng, Y. Decreased androgen receptor expression is associated with distant metastases in patients with androgen receptor–expressing triple-negative breast carcinoma. Am. J. Clin. Pathol. 2012, 138, 511–516. [Google Scholar] [CrossRef]
- Tang, D.; Xu, S.; Zhang, Q.; Zhao, W. The expression and clinical significance of the androgen receptor and E-cadherin in triple-negative breast cancer. Med. Oncol. 2012, 29, 526–533. [Google Scholar] [CrossRef]
- Yue, Y.; Astvatsaturyan, K.; Cui, X.; Zhang, X.; Fraass, B.; Bose, S. Stratification of prognosis of triple-negative breast cancer patients using combinatorial biomarkers. PLoS ONE 2016, 11, e0149661. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Peng, R.; Yuan, Z.; Wang, S.; Peng, J.; Lin, G.; Jiang, X.; Qin, T. Prognostic value of androgen receptor expression in operable triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med. Oncol. 2012, 29, 406–410. [Google Scholar] [CrossRef]
- Robinson, J.L.; MacArthur, S.; Ross-Innes, C.S.; Tilley, W.D.; Neal, D.E.; Mills, I.G.; Carroll, J.S. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 2012, 31, 1617. [Google Scholar] [CrossRef]
- Mrklić, I.; Pogorelić, Z.; Ćapkun, V.; Tomić, S. Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem. 2013, 115, 344–348. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- Qu, Q.; Mao, Y.; Fei, X.-C.; Shen, K.-W. The impact of androgen receptor expression on breast cancer survival: A retrospective study and meta-analysis. PLoS ONE 2013, 8, e82650. [Google Scholar] [CrossRef]
- Wang, C.; Pan, B.; Zhu, H.; Zhou, Y.; Mao, F.; Lin, Y.; Xu, Q.; Sun, Q. Prognostic value of androgen receptor in triple negative breast cancer: A meta-analysis. Oncotarget 2016, 7, 46482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Palla, S.L.; Carey, M.; Agarwal, R.; Meric-Berstam, F.; Traina, T.A.; Hudis, C.; Hortobagyi, G.N.; Gerald, W.L. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin. Cancer Res. 2009, 15, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Shi, Y.; Li, Z.; Jiang, W. Expression and clinical significance of androgen receptor in triple negative breast cancer. Chin. J. Cancer 2010, 29, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Kashiwagi, S.; Goto, W.; Tanaka, S.; Morisaki, T.; Takashima, T.; Noda, S.; Onoda, N.; Ohsawa, M.; Hirakawa, K.; et al. Expression and Clinical Significance of Androgen Receptor in Triple-Negative Breast Cancer. Cancers 2017, 9, 4. [Google Scholar] [CrossRef]
- McGhan, L.J.; McCullough, A.E.; Protheroe, C.A.; Dueck, A.C.; Lee, J.J.; Nunez-Nateras, R.; Castle, E.P.; Gray, R.J.; Wasif, N.; Goetz, M.P. Androgen receptor-positive triple negative breast cancer: A unique breast cancer subtype. Ann. Surg. Oncol. 2014, 21, 361–367. [Google Scholar] [CrossRef]
- Choi, J.E.; Kang, S.H.; Lee, S.J.; Bae, Y.K. Androgen receptor expression predicts decreased survival in early stage triple-negative breast cancer. Ann. Surg. Oncol. 2015, 22, 82–89. [Google Scholar] [CrossRef]
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011, 17, 1867–1874. [Google Scholar] [CrossRef]
- Vera-Badillo, F.E.; Templeton, A.J.; de Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen receptor expression and outcomes in early breast cancer: A systematic review and meta-analysis. JNCI J. Natl. Cancer Inst. 2014, 106, djt319. [Google Scholar] [CrossRef]
- Jiagge, E.; Jibril, A.S.; Davis, M.; Murga-Zamalloa, C.; Kleer, C.G.; Gyan, K.; Divine, G.; Hoenerhoff, M.; Bensenhave, J.; Awuah, B.; et al. Androgen Receptor and ALDH1 Expression Among Internationally Diverse Patient Populations. J. Glob. Oncol. 2018, 4, 1–8. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS ONE 2013, 8, e77060. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Pollinger, B.; Ohmer, M.; Hofmann, F.; Nigg, E.A.; Hemmings, B.A.; Wartmann, M. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 2012, 125, 5391–5402. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef]
- Zhao, J.Z.; Ye, Q.; Wang, L.; Lee, S.C. Centrosome amplification in cancer and cancer-associated human diseases. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188566. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.X.; Sperry, A.O. C-terminal kinesin motor KIFC1 participates in acrosome biogenesis and vesicle transport. Biol. Reprod. 2003, 69, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mikule, K.; Wang, W.; Su, N.; Petteruti, P.; Gharahdaghi, F.; Code, E.; Zhu, X.; Jacques, K.; Lai, Z.; et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem. Biol. 2013, 8, 2201–2208. [Google Scholar] [CrossRef] [PubMed]
- Pannu, V.; Rida, P.C.; Ogden, A.; Turaga, R.C.; Donthamsetty, S.; Bowen, N.J.; Rudd, K.; Gupta, M.V.; Reid, M.D.; Cantuaria, G.; et al. HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients. Oncotarget 2015, 6, 6076–6091. [Google Scholar] [CrossRef]
- Patel, N.; Weekes, D.; Drosopoulos, K.; Gazinska, P.; Noel, E.; Rashid, M.; Mirza, H.; Quist, J.; Braso-Maristany, F.; Mathew, S.; et al. Integrated genomics and functional validation identifies malignant cell specific dependencies in triple negative breast cancer. Nat. Commun. 2018, 9, 1044. [Google Scholar] [CrossRef]
- De, S.; Cipriano, R.; Jackson, M.W.; Stark, G.R. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res. 2009, 69, 8035–8042. [Google Scholar] [CrossRef]
- Ogden, A.; Garlapati, C.; Li, X.B.; Turaga, R.C.; Oprea-Ilies, G.; Wright, N.; Bhattarai, S.; Mittal, K.; Wetherilt, C.S.; Krishnamurti, U.; et al. Multi-institutional study of nuclear KIFC1 as a biomarker of poor prognosis in African American women with triple-negative breast cancer. Sci. Rep. 2017, 7, 42289. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, W.; Chen, D.; Boohaker, R.J.; Zhai, L.; Padmalayam, I.; Wennerberg, K.; Xu, B.; Zhang, W. KIFC1 is a novel potential therapeutic target for breast cancer. Cancer Biol. Ther. 2015, 16, 1316–1322. [Google Scholar] [CrossRef]
- Mehrotra, J.; Ganpat, M.M.; Kanaan, Y.; Fackler, M.J.; McVeigh, M.; Lahti-Domenici, J.; Polyak, K.; Argani, P.; Naab, T.; Garrett, E.; et al. Estrogen receptor/progesterone receptor-negative breast cancers of young African-American women have a higher frequency of methylation of multiple genes than those of Caucasian women. Clin. Cancer Res. 2004, 10, 2052–2057. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Dorsey, T.H.; Terunuma, A.; Kittles, R.A.; Ambs, S.; Kwabi-Addo, B. Relationship between tumor DNA methylation status and patient characteristics in African-American and European-American women with breast cancer. PLoS ONE 2012, 7, e37928. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Edmiston, S.N.; Tse, C.K.; Bryant, C.; Kuan, P.F.; Hair, B.Y.; Parrish, E.A.; May, R.; Swift-Scanlan, T. Racial variation in breast tumor promoter methylation in the Carolina Breast Cancer Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Ambrosone, C.B.; Young, A.C.; Sucheston, L.E.; Wang, D.; Yan, L.; Liu, S.; Tang, L.; Hu, Q.; Freudenheim, J.L.; Shields, P.G.; et al. Genome-wide methylation patterns provide insight into differences in breast tumor biology between American women of African and European ancestry. Oncotarget 2014, 5, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Garlapati, C.; Aneja, R. Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations. Cancers 2022, 14, 1903. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. The Epigenetic Landscape of Promoter Genome-wide Analysis in Breast Cancer. Sci. Rep. 2017, 7, 6597. [Google Scholar] [CrossRef]
- Muhammad, A.; Forcados, G.E.; Katsayal, B.S.; Bako, R.S.; Aminu, S.; Sadiq, I.Z.; Abubakar, M.B.; Yusuf, A.P.; Malami, I.; Faruk, M.; et al. Potential epigenetic modifications implicated in triple- to quadruple-negative breast cancer transition: A review. Epigenomics 2022, 14, 711–726. [Google Scholar] [CrossRef]
- Qattan, A.; Al-Tweigeri, T.; Suleman, K. Translational Implications of Dysregulated Pathways and microRNA Regulation in Quadruple-Negative Breast Cancer. Biomedicines 2022, 10, 366. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, Z. MicroRNA-432 Acts as a Prognostic Biomarker and an Inhibitor of Cell Proliferation, Migration, and Invasion in Breast Cancer. Clin. Breast Cancer 2021, 21, e462–e470. [Google Scholar] [CrossRef]
- Dattilo, M.A.; Benzo, Y.; Herrera, L.M.; Prada, J.G.; Castillo, A.F.; Orlando, U.D.; Podesta, E.J.; Maloberti, P.M. Regulatory mechanisms leading to differential Acyl-CoA synthetase 4 expression in breast cancer cells. Sci. Rep. 2019, 9, 10324. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, J.; Lin, Y.; Sun, X.M.; Zhang, J.N.; Cheng, Z.Q. Identification of MiR-211-5p as a tumor suppressor by targeting ACSL4 in Hepatocellular Carcinoma. J. Transl. Med. 2020, 18, 326. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Zhang, S.; Shan, C.; Zhou, L.; Zhou, Z. microRNA-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the EGFR/Akt signaling pathway. FEBS J. 2013, 280, 3962–3974. [Google Scholar] [CrossRef] [PubMed]
- Uva, P.; Cossu-Rocca, P.; Loi, F.; Pira, G.; Murgia, L.; Orru, S.; Floris, M.; Muroni, M.R.; Sanges, F.; Carru, C.; et al. miRNA-135b Contributes to Triple Negative Breast Cancer Molecular Heterogeneity: Different Expression Profile in Basal-like Versus non-Basal-like Phenotypes. Int. J. Med. Sci. 2018, 15, 536–548. [Google Scholar] [CrossRef]
- Yao, S.; Graham, K.; Shen, J.; Campbell, L.E.; Singh, P.; Zirpoli, G.; Roberts, M.; Ciupak, G.; Davis, W.; Hwang, H.; et al. Genetic variants in microRNAs and breast cancer risk in African American and European American women. Breast Cancer Res. Treat. 2013, 141, 447–459. [Google Scholar] [CrossRef]
- Wright, N.; Akinyemiju, T.; Subhedar, P.; Rida, P.; Aneja, R. Targeting risk factors for reducing the racially disparate burden in breast cancer. Front. Biosci. (Schol. Ed.) 2019, 11, 136–160. [Google Scholar] [CrossRef] [PubMed]
- Kantor, O.; Wang, M.L.; Bertrand, K.; Pierce, L.; Freedman, R.A.; Chavez-MacGregor, M.; King, T.A.; Mittendorf, E.A. Racial and Socioeconomic Disparities in Breast Cancer Outcomes within the AJCC Pathologic Prognostic Staging System. Ann. Surg. Oncol. 2022, 29, 686–696. [Google Scholar] [CrossRef]
- Siddharth, S.; Sharma, D. Racial Disparity and Triple-Negative Breast Cancer in African-American Women: A Multifaceted Affair between Obesity, Biology, and Socioeconomic Determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef]
- Martini, R.; Newman, L.; Davis, M. Breast cancer disparities in outcomes; unmasking biological determinants associated with racial and genetic diversity. Clin. Exp. Metastasis 2022, 39, 7–14. [Google Scholar] [CrossRef]
- Black, S.A. Diabetes, diversity, and disparity: What do we do with the evidence? Am. J. Public Health 2002, 92, 543–548. [Google Scholar] [CrossRef]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef]
- Kelsey, J.L.; Gammon, M.D.; John, E.M. Reproductive factors and breast cancer. Epidemiol. Rev. 1993, 15, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Forshee, R.A.; Storey, M.L.; Ritenbaugh, C. Breast cancer risk and lifestyle differences among premenopausal and postmenopausal African-American women and white women. Cancer 2003, 97, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Monk, J.M.; Robinson, L.E.; Mourtzakis, M. The integrative role of leptin, oestrogen and the insulin family in obesity-associated breast cancer: Potential effects of exercise. Obes. Rev. 2015, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.R.; O’Brien, J.; Borges, V.F.; Conklin, M.W.; Keely, P.J.; Eliceiri, K.W.; Marusyk, A.; Tan, A.C.; Schedin, P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat. Med. 2011, 17, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Basree, M.M.; Shinde, N.; Koivisto, C.; Cuitino, M.; Kladney, R.; Zhang, J.; Stephens, J.; Palettas, M.; Zhang, A.; Kim, H.K.; et al. Abrupt involution induces inflammation, estrogenic signaling, and hyperplasia linking lack of breastfeeding with increased risk of breast cancer. Breast Cancer Res. 2019, 21, 80. [Google Scholar] [CrossRef] [PubMed]
- Crosswell, A.D.; Bower, J.E.; Ganz, P.A. Childhood adversity and inflammation in breast cancer survivors. Psychosom. Med. 2014, 76, 208–214. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jinna, N.; Jovanovic-Talisman, T.; LaBarge, M.; Natarajan, R.; Kittles, R.; Sistrunk, C.; Rida, P.; Seewaldt, V.L. Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention. Cancers 2022, 14, 4484. https://doi.org/10.3390/cancers14184484
Jinna N, Jovanovic-Talisman T, LaBarge M, Natarajan R, Kittles R, Sistrunk C, Rida P, Seewaldt VL. Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention. Cancers. 2022; 14(18):4484. https://doi.org/10.3390/cancers14184484
Chicago/Turabian StyleJinna, Nikita, Tijana Jovanovic-Talisman, Mark LaBarge, Rama Natarajan, Rick Kittles, Christopher Sistrunk, Padmashree Rida, and Victoria L. Seewaldt. 2022. "Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention" Cancers 14, no. 18: 4484. https://doi.org/10.3390/cancers14184484
APA StyleJinna, N., Jovanovic-Talisman, T., LaBarge, M., Natarajan, R., Kittles, R., Sistrunk, C., Rida, P., & Seewaldt, V. L. (2022). Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention. Cancers, 14(18), 4484. https://doi.org/10.3390/cancers14184484