
Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance

 , , , , and
, , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
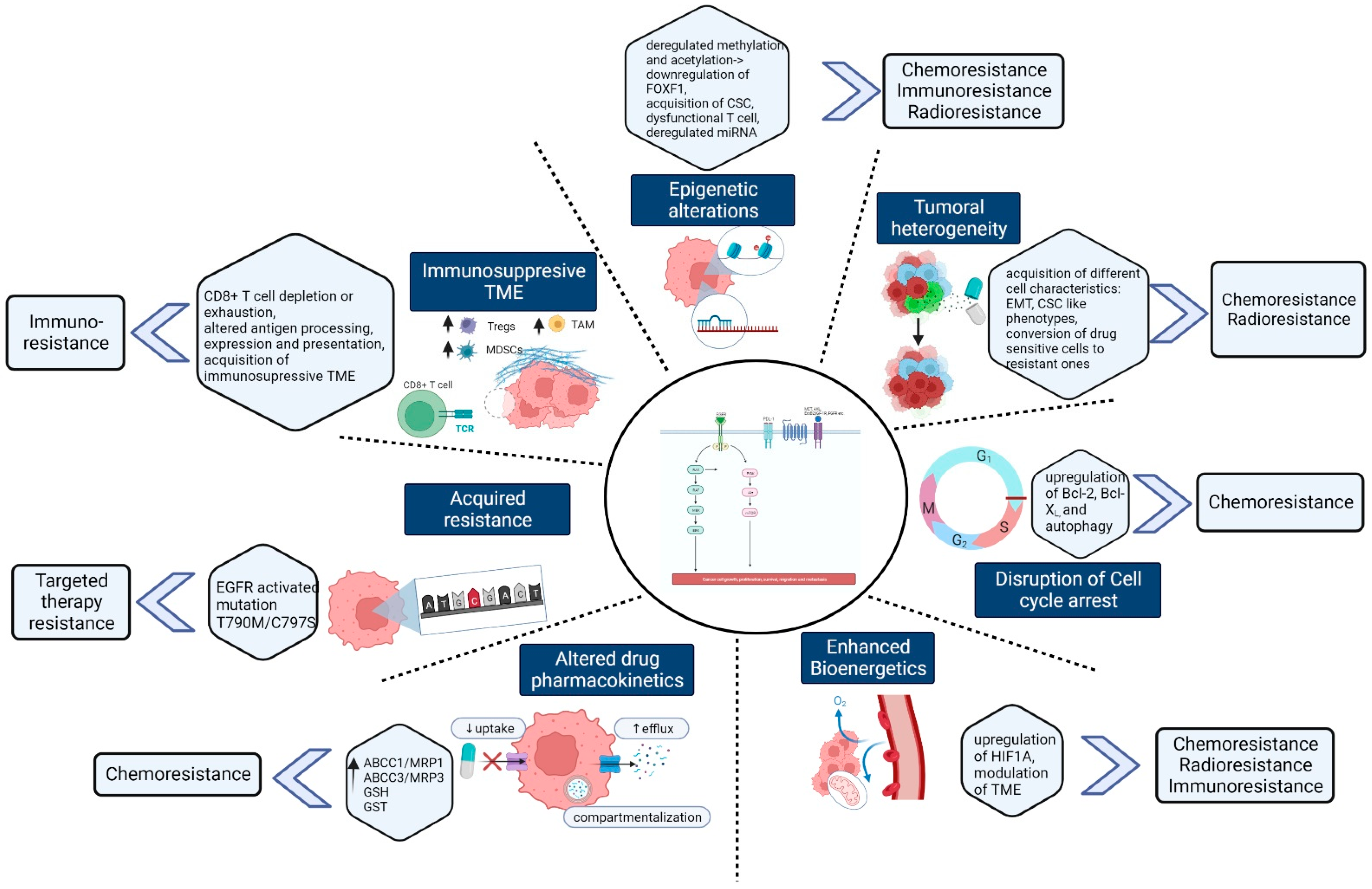
2. Mechanism of Chemoresistance and Potential Therapeutic Inhibitors
3. Mechanism of Radiotherapy Resistance in Lung Cancer
3.1. Targeting Signaling Pathways Associated with Radioresistance and Potential Strategies for Radiosensitization in Lung Cancer
3.2. miRNAs in Radioresistant Lung Cancer Cells
3.3. DNA Damage Associated with Radioresistance in Lung Cancer
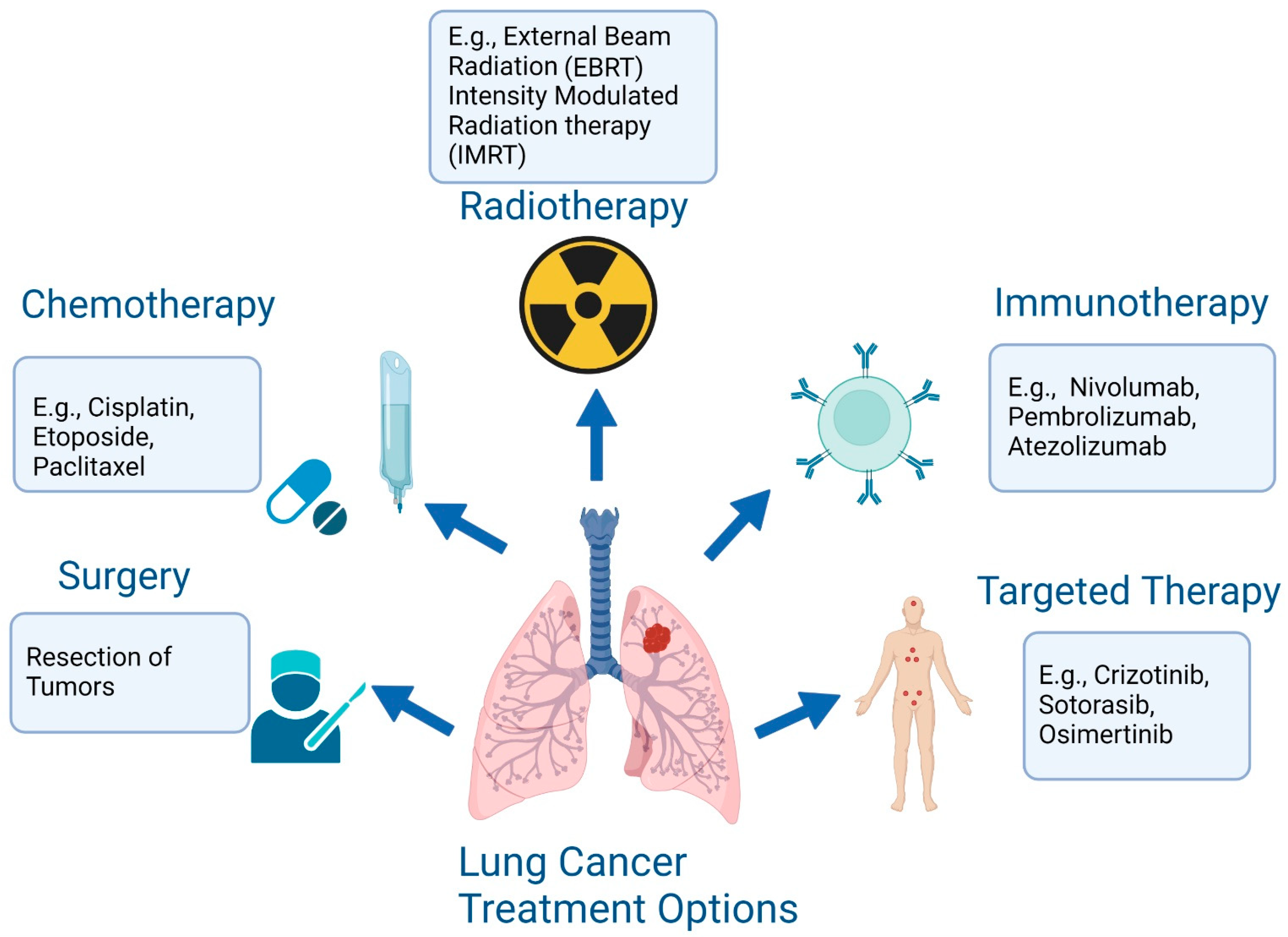
4. Targeted Therapy Resistance in Lung cancer and Overcoming Strategies
4.1. Overcoming EGFR TKI Resistance
4.2. Overcoming ALK Drug Resistance
4.3. Strategies to Overcome Resistance to KRAS G12C Inhibitors
5. Mechanism of Immunotherapeutic Resistance and Overcoming Strategies
Targeting Immunoresistance Mechanism to Resensitize Lung Cancer Cells to Immunotherapy
6. Combination Approaches for Improving Therapeutic Resistance and Future Prospects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Współczesna Onkol. 2021, 25, 45–52. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Zhao, W.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; Zhang, T.; Zhu, B.; Wang, M.; Jones, K.; Hicks, B.; et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat. Commun. 2020, 11, 2459. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Pao, W.; Hutchinson, K.E. Chipping away at the lung cancer genome. Nat. Med. 2012, 18, 349–351. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Saab, S.; Zalzale, H.; Rahal, Z.; Khalifeh, Y.; Sinjab, A.; Kadara, H. Insights Into Lung Cancer Immune-Based Biology, Prevention, and Treatment. Front. Immunol. 2020, 11, 159. [Google Scholar] [CrossRef]
- Hung, Y.P.; Chirieac, L.R. How should molecular findings be integrated in the classification for lung cancer? Transl. Lung Cancer Res. 2020, 9, 2245–2254. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, Q.; Huang, Z.; Li, J. Combining immunotherapy and radiotherapy in lung cancer: A promising future? J. Thorac. Dis. 2020, 12, 4498–4503. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef]
- Jain, P.; Jain, C.; Velcheti, V. Role of immune-checkpoint inhibitors in lung cancer. Ther. Adv. Respir. Dis. 2018, 12, 175346581775007. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Min, H.-Y.; Lee, H.-Y. Mechanisms of resistance to chemotherapy in non-small cell lung cancer. Arch. Pharmacal Res. 2021, 44, 146–164. [Google Scholar] [CrossRef]
- Janet, W.-T.; Elizabeth, H.-B. Drug Resistance Mechanisms in Non-Small Cell Lung Carcinoma. J. Cancer Res. Updates 2013, 2, 265. [Google Scholar] [CrossRef]
- Konstantinov, S.M.; Berger, M.R. Alkylating Agents. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 53–57. [Google Scholar]
- Olaussen, K.A.; Postel-Vinay, S. Predictors of chemotherapy efficacy in non-small-cell lung cancer: A challenging landscape. Ann. Oncol. 2016, 27, 2004–2016. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Selvaggi, G. Antimetabolites and cancer: Emerging data with a focus on antifolates. Expert Opin. Ther. Pat. 2006, 16, 189–200. [Google Scholar] [CrossRef]
- Riganti, C.; Contino, M. New Strategies to Overcome Resistance to Chemotherapy and Immune System in Cancer. Int. J. Mol. Sci. 2019, 20, 4783. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Chen, P.; Kuang, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; Yu, F.; et al. Mechanisms of drugs-resistance in small cell lung cancer: DNA-related, RNA-related, apoptosis-related, drug accumulation and metabolism procedure. Transl. Lung Cancer Res. 2020, 9, 768–786. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, J.-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. [Google Scholar] [CrossRef]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum Resistance: The Role of DNA Repair Pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef]
- Vilmar, A.; Sørensen, J.B. Excision repair cross-complementation group 1 (ERCC1) in platinum-based treatment of non-small cell lung cancer with special emphasis on carboplatin: A review of current literature. Lung Cancer 2009, 64, 131–139. [Google Scholar] [CrossRef]
- Young, L.C.; Campling, B.G.; Cole, S.P.; Deeley, R.G.; Gerlach, J.H. Multidrug resistance proteins MRP3, MRP1, and MRP2 in lung cancer: Correlation of protein levels with drug response and messenger RNA levels. Clin. Cancer Res. 2001, 7, 1798–1804. [Google Scholar]
- Oguri, T.; Ozasa, H.; Uemura, T.; Bessho, Y.; Miyazaki, M.; Maeno, K.; Maeda, H.; Sato, S.; Ueda, R. MRP7/ABCC10 expression is a predictive biomarker for the resistance to paclitaxel in non-small cell lung cancer. Mol. Cancer Ther. 2008, 7, 1150–1155. [Google Scholar] [CrossRef]
- Bessho, Y.; Oguri, T.; Ozasa, H.; Uemura, T.; Sakamoto, H.; Miyazaki, M.; Maeno, K.; Sato, S.; Ueda, R. ABCC10/MRP7 is associated with vinorelbine resistance in non-small cell lung cancer. Oncol. Rep. 2009, 21, 263–268. [Google Scholar]
- Shimomura, M.; Yaoi, T.; Itoh, K.; Kato, D.; Terauchi, K.; Shimada, J.; Fushiki, S. Drug resistance to paclitaxel is not only associated with ABCB1 mRNA expression but also with drug accumulation in intracellular compartments in human lung cancer cell lines. Int. J. Oncol. 2012, 40, 995–1004. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, H.; Yan, A.; Yang, Y.; Meng, Q.; Sun, L.; Pang, H.; Li, C.; Dong, X.; Cai, L. ABCC3 as a marker for multidrug resistance in non-small cell lung cancer. Sci. Rep. 2013, 3, 3120. [Google Scholar] [CrossRef]
- Shen, W.; Pang, H.; Liu, J.; Zhou, J.; Zhang, F.; Liu, L.; Ma, N.; Zhang, N.; Zhang, H.; Liu, L. Epithelial-mesenchymal transition contributes to docetaxel resistance in human non-small cell lung cancer. Oncol. Res. 2014, 22, 47–55. [Google Scholar] [CrossRef]
- Cui, H.; Arnst, K.; Miller, D.D.; Li, W. Recent Advances in Elucidating Paclitaxel Resistance Mechanisms in Non-small Cell Lung Cancer and Strategies to Overcome Drug Resistance. Curr. Med. Chem. 2020, 27, 6573–6595. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Sharma, A.; Patrick, B.; Li, J.; Sharma, R.; Jeyabal, P.V.; Reddy, P.M.; Awasthi, S.; Awasthi, Y.C. Glutathione S-transferases as antioxidant enzymes: Small cell lung cancer (H69) cells transfected with hGSTA1 resist doxorubicin-induced apoptosis. Arch. Biochem. Biophys. 2006, 452, 165–173. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Nie, J.; Huang, J.-P.; Zheng, G.-J.; Feng, B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed. Pharmacother. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chun, J.; Pan, C.; Li, D.; Lin, R.; Alesi, G.N.; Wang, X.; Kang, H.B.; Song, L.; Wang, D.; et al. MAST1 Drives Cisplatin Resistance in Human Cancers by Rewiring cRaf-Independent MEK Activation. Cancer Cell 2018, 34, 315–330.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol./Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shen, M.; Yang, L.; Yang, X.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Simultaneous targeting of ATM and Mcl-1 increases cisplatin sensitivity of cisplatin-resistant non-small cell lung cancer. Cancer Biol. Ther. 2017, 18, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Horibe, S.; Matsuda, A.; Tanahashi, T.; Inoue, J.; Kawauchi, S.; Mizuno, S.; Ueno, M.; Takahashi, K.; Maeda, Y.; Maegouchi, T.; et al. Cisplatin resistance in human lung cancer cells is linked with dysregulation of cell cycle associated proteins. Life Sci. 2015, 124, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef]
- Shen, M.; Xu, Z.; Xu, W.; Jiang, K.; Zhang, F.; Ding, Q.; Xu, Z.; Chen, Y. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 149. [Google Scholar] [CrossRef]
- Hu, Y.; Yagüe, E.; Zhao, J.; Wang, L.; Bai, J.; Yang, Q.; Pan, T.; Zhao, H.; Liu, J.; Zhang, J. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018, 423, 47–59. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J.; et al. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27 (Suppl. S1), S149–S157. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Targets. 2013, 17, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodur, C.; Basaga, H. Bcl-2 inhibitors: Emerging drugs in cancer therapy. Curr. Med. Chem. 2012, 19, 1804–1820. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Chapter Three—Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 137, pp. 37–75. [Google Scholar]
- Inno, A.; Stagno, A.; Gori, S. Schlafen-11 (SLFN11): A step forward towards personalized medicine in small-cell lung cancer? Transl. Lung Cancer Res. 2018, 7 (Suppl. S4), S341–S345. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-H.; Zhao, X.; Zhu, J.; Kim, I.-K.; Rao, G.; McCutcheon, J.; Hsu, S.-T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Checkpoint Kinase 1 Inhibition Enhances Cisplatin Cytotoxicity and Overcomes Cisplatin Resistance in SCLC by Promoting Mitotic Cell Death. J. Thorac. Oncol. 2019, 14, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Sosa Iglesias, V.; Giuranno, L.; Dubois, L.J.; Theys, J.; Vooijs, M. Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting? Front. Oncol. 2018, 8, 267. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Minari, R.; Cortellini, A.; Rolfo, C.D.; Giovannetti, E.; Tiseo, M. Notch pathway in small-cell lung cancer: From preclinical evidence to therapeutic challenges. Cell Oncol. 2019, 42, 261–273. [Google Scholar] [CrossRef]
- Geisslinger, F.; Müller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance-A Mini Review. Front. Oncol. 2020, 10, 1156. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, K.; Li, Q.; Zou, Y.; Chen, B.; Gong, Q.; Ho, H.I.; Yin, T.; Zhang, F.; Lu, Y.; et al. The Novel Autophagy Inhibitor Alpha-Hederin Promoted Paclitaxel Cytotoxicity by Increasing Reactive Oxygen Species Accumulation in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3221. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wu, Q.; Zhou, X.; Huang, J. SphK1 functions downstream of IGF-1 to modulate IGF-1-induced EMT, migration and paclitaxel resistance of A549 cells: A preliminary in vitro study. J. Cancer 2019, 10, 4264–4269. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Zhao, J.; Xue, X.; Fu, W.; Dai, L.; Jiang, Z.; Zhong, S.; Deng, B.; Yin, J. Epigenetic activation of FOXF1 confers cancer stem cell properties to cisplatin-resistant non-small cell lung cancer. Int. J. Oncol. 2020, 56, 1083–1092. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Reshma, P.L.; Unnikrishnan, B.S.; Preethi, G.U.; Syama, H.P.; Archana, M.G.; Remya, K.; Shiji, R.; Sreekutty, J.; Sreelekha, T.T. Overcoming drug-resistance in lung cancer cells by paclitaxel loaded galactoxyloglucan nanoparticles. Int. J. Biol. Macromol. 2019, 136, 266–274. [Google Scholar] [CrossRef]
- Pearce, M.C.; Gamble, J.T.; Kopparapu, P.R.; O’Donnell, E.F.; Mueller, M.J.; Jang, H.S.; Greenwood, J.A.; Satterthwait, A.C.; Tanguay, R.L.; Zhang, X.-K.; et al. Induction of apoptosis and suppression of tumor growth by Nur77-derived Bcl-2 converting peptide in chemoresistant lung cancer cells. Oncotarget 2018, 9, 26072–26085. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, B.; Wang, Q.; Ye, M.; Qiu, Q.; Zhou, Y.; Zeng, F.; Zhang, X.; Guo, Y.; Guo, L. Long non-coding RNA HOTTIP promotes BCL-2 expression and induces chemoresistance in small cell lung cancer by sponging miR-216a. Cell Death Dis. 2018, 9, 85. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, H.; Xue, R.; Tang, W.; Zhang, S. BH3 Mimetic ABT-199 Enhances the Sensitivity of Gemcitabine in Pancreatic Cancer in vitro and in vivo. Dig. Dis. Sci. 2018, 63, 3367–3375. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, K.; Wu, J.; Shi, J.; Xue, J.; Li, J.; Chen, J.; Zhu, Y.; Wei, J.; He, J.; et al. Wnt5a Increases Properties of Lung Cancer Stem Cells and Resistance to Cisplatin through Activation of Wnt5a/PKC Signaling Pathway. Stem Cells Int. 2016, 2016, 1690896. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; De Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J.-C.; et al. Different Levels of Twist1 Regulate Skin Tumor Initiation, Stemness, and Progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Yang, W.-H.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Tzeng, C.-H.; Kao, S.-Y.; Wu, K.-J.; Hung, M.-C.; Yang, M.-H. RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef]
- Herzog, B.H.; Devarakonda, S.; Govindan, R. Overcoming Chemotherapy Resistance in SCLC. J. Thorac. Oncol. 2021, 16, 2002–2015. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef]
- Hua, L.; Zhu, G.; Wei, J. MicroRNA-1 overexpression increases chemosensitivity of non-small cell lung cancer cells by inhibiting autophagy related 3-mediated autophagy. Cell Biol. Int. 2018, 42, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagryazhskaya, A.; Zhivotovsky, B. miRNAs in lung cancer: A link to aging. Ageing Res. Rev. 2014, 17, 54–67. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Zhang, S.; Zheng, X.; Xie, S.; Mao, J.; Cai, Y.; Lu, X.; Hu, L.; Shen, J.; et al. MiR-223 regulates autophagy associated with cisplatin resistance by targeting FBXW7 in human non-small cell lung cancer. Cancer Cell Int. 2020, 20, 258. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Finn, S.P.; Cuffe, S.; O’Byrne, K.J.; Barr, M.P. The emerging role of microRNAs in resistance to lung cancer treatments. Cancer Treat. Rev. 2015, 41, 160–169. [Google Scholar] [CrossRef]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepeñas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Obrist, F.; Michels, J.; Durand, S.; Chery, A.; Pol, J.; Levesque, S.; Joseph, A.; Astesana, V.; Pietrocola, F.; Wu, G.S.; et al. Metabolic vulnerability of cisplatin-resistant cancers. EMBO J. 2018, 37, e98597. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 2019, 18, 69. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef] [Green Version]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The Role of Eukaryotic and Prokaryotic ABC Transporter Family in Failure of Chemotherapy. Front. Pharm. 2016, 7, 535. [Google Scholar] [CrossRef]
- Xu, X.; Jin, S.; Ma, Y.; Fan, Z.; Yan, Z.; Li, W.; Song, Q.; You, W.; Lyu, Z.; Song, Y.; et al. miR-30a-5p enhances paclitaxel sensitivity in non-small cell lung cancer through targeting BCL-2 expression. J. Mol. Med. 2017, 95, 861–871. [Google Scholar] [CrossRef]
- Ibanez de Caceres, I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguán-García, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; de CastroCarpeño, J.; et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef]
- Dalvi, M.P.; Wang, L.; Zhong, R.; Kollipara, R.K.; Park, H.; Bayo, J.; Yenerall, P.; Zhou, Y.; Timmons, B.C.; Rodriguez-Canales, J.; et al. Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors. Cell Rep. 2017, 19, 1669–1684. [Google Scholar] [CrossRef]
- Chen, B.; Tan, Y.; Liang, Y.; Li, Y.; Chen, L.; Wu, S.; Xu, W.; Wang, Y.; Zhao, W.; Wu, J. Per2 participates in AKT-mediated drug resistance in A549/DDP lung adenocarcinoma cells. Oncol. Lett. 2017, 13, 423–428. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem–like Cells in Human Non–Small Cell Lung Cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef]
- Ahmad, A.; Maitah, M.i.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef]
- Wu, H.-M.; Jiang, Z.-F.; Ding, P.-S.; Shao, L.-J.; Liu, R.-Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Guo, Q.; Lan, F.; Yan, X.; Xiao, Z.; Wu, Y.; Zhang, Q. Hypoxia exposure induced cisplatin resistance partially via activating p53 and hypoxia inducible factor-1α in non-small cell lung cancer A549 cells. Oncol. Lett. 2018, 16, 801–808. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef]
- Gomez-Casal, R.; Epperly, M.W.; Wang, H.; Proia, D.A.; Greenberger, J.S.; Levina, V. Radioresistant human lung adenocarcinoma cells that survived multiple fractions of ionizing radiation are sensitive to HSP90 inhibition. Oncotarget 2015, 6, 44306–44322. [Google Scholar] [CrossRef]
- Yang, W.-C.; Hsu, F.-M.; Yang, P.-C. Precision radiotherapy for non-small cell lung cancer. J. Biomed. Sci. 2020, 27, 82. [Google Scholar] [CrossRef]
- Chen, M.; Yang, J.; Liao, Z.; Chen, J.; Xu, C.; He, X.; Zhang, X.; Zhu, R.X.; Li, H. Anatomic change over the course of treatment for non–small cell lung cancer patients and its impact on intensity-modulated radiation therapy and passive-scattering proton therapy deliveries. Radiat. Oncol. 2020, 15, 55. [Google Scholar] [CrossRef]
- Kim, N.; Noh, J.M.; Lee, W.; Park, B.; Pyo, H. Clinical Outcomes of Pencil Beam Scanning Proton Therapy in Locally Advanced Non-Small Cell Lung Cancer: Propensity Score Analysis. Cancers 2021, 13, 3497. [Google Scholar] [CrossRef]
- Skliarenko, J.; Barry, A. Clinical and practical applications of radiation therapy: When should radiation therapy be considered for my patient? Medicine 2020, 48, 84–89. [Google Scholar] [CrossRef]
- Cao, C.; Wang, D.; Chung, C.; Tian, D.; Rimner, A.; Huang, J.; Jones, D.R. A systematic review and meta-analysis of stereotactic body radiation therapy versus surgery for patients with non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2019, 157, 362–373.e8. [Google Scholar] [CrossRef]
- Yin, X.; Yan, D.; Qiu, M.; Huang, L.; Yan, S.-X. Prophylactic cranial irradiation in small cell lung cancer: A systematic review and meta-analysis. BMC Cancer 2019, 19, 95. [Google Scholar] [CrossRef]
- Chun, S.G.; Hu, C.; Choy, H.; Komaki, R.U.; Timmerman, R.D.; Schild, S.E.; Bogart, J.A.; Dobelbower, M.C.; Bosch, W.; Galvin, J.M.; et al. Impact of Intensity-Modulated Radiation Therapy Technique for Locally Advanced Non-Small-Cell Lung Cancer: A Secondary Analysis of the NRG Oncology RTOG 0617 Randomized Clinical Trial. J. Clin. Oncol. 2017, 35, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Liao, Z. Proton therapy for locally advanced non-small cell lung cancer. Br. J. Radiol. 2020, 93, 20190378. [Google Scholar] [CrossRef]
- Iocolano, M.; Wild, A.T.; Hannum, M.; Zhang, Z.; Simone, C.B., 2nd; Gelblum, D.; Wu, A.J.; Rimner, A.; Shepherd, A.F. Hypofractionated vs. conventional radiation therapy for stage III non-small cell lung cancer treated without chemotherapy. Acta Oncol. 2020, 59, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Xue, J.; Li, R.; Zhou, L.; Deng, L.; Chen, L.; Zhang, Y.; Li, Y.; Zhang, X.; Xiu, W.; et al. Effect of Low-Dose Radiation Therapy on Abscopal Responses to Hypofractionated Radiation Therapy and Anti-PD1 in Mice and Patients With Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Yi, P.; Wang, H.; Xia, L.; Han, Y.; Wang, H.; Zeng, B.; Tang, L.; Pan, Q.; Tian, Y.; et al. RAC1 Involves in the Radioresistance by Mediating Epithelial-Mesenchymal Transition in Lung Cancer. Front. Oncol. 2020, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Toulany, M.; Iida, M.; Keinath, S.; Iyi, F.F.; Mueck, K.; Fehrenbacher, B.; Mansour, W.Y.; Schaller, M.; Wheeler, D.L.; Rodemann, H.P. Dual targeting of PI3K and MEK enhances the radiation response of K-RAS mutated non-small cell lung cancer. Oncotarget 2016, 7, 43746–43761. [Google Scholar] [CrossRef]
- Zhao, R.S.; Wang, W.; Li, J.P.; Liu, C.M.; He, L. Gelsolin Promotes Radioresistance in Non-Small Cell Lung Cancer Cells Through Activation of Phosphoinositide 3-Kinase/Akt Signaling. Technol. Cancer Res. Treat. 2017, 16, 512–518. [Google Scholar] [CrossRef]
- Li, L.; Li, Y.; Zou, H. A novel role for apatinib in enhancing radiosensitivity in non-small cell lung cancer cells by suppressing the AKT and ERK pathways. PeerJ 2021, 9, e12356. [Google Scholar] [CrossRef]
- Ushijima, H.; Suzuki, Y.; Oike, T.; Komachi, M.; Yoshimoto, Y.; Ando, K.; Okonogi, N.; Sato, H.; Noda, S.E.; Saito, J.; et al. Radio-sensitization effect of an mTOR inhibitor, temsirolimus, on lung adenocarcinoma A549 cells under normoxic and hypoxic conditions. J. Radiat. Res. 2015, 56, 663–668. [Google Scholar] [CrossRef]
- Das, U.; Manna, K.; Adhikary, A.; Mishra, S.; Saha, K.D.; Sharma, R.D.; Majumder, B.; Dey, S. Ferulic acid enhances the radiation sensitivity of lung and liver carcinoma cells by collapsing redox homeostasis: Mechanistic involvement of Akt/p38 MAPK signalling pathway. Free Radic. Res. 2019, 53, 944–967. [Google Scholar] [CrossRef]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. 2021, 48, 238–245. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, D.W.; Kang, C.M.; Hong, S.H. A novel PPARɣ ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells. Exp. Mol. Med. 2020, 52, 1730–1743. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mikac, S.; Rychłowski, M.; Dziadosz, A.; Szabelska-Beresewicz, A.; Fahraeus, R.; Hupp, T.; Sznarkowska, A. Identification of a Stable, Non-Canonically Regulated Nrf2 Form in Lung Cancer Cells. Antioxidants 2021, 10, 786. [Google Scholar] [CrossRef]
- Binkley, M.S.; Jeon, Y.J.; Nesselbush, M.; Moding, E.J.; Nabet, B.Y.; Almanza, D.; Kunder, C.; Stehr, H.; Yoo, C.H.; Rhee, S.; et al. KEAP1/NFE2L2 Mutations Predict Lung Cancer Radiation Resistance That Can Be Targeted by Glutaminase Inhibition. Cancer Discov. 2020, 10, 1826–1841. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhang, M.; Zhou, C.; Wang, W.; Yang, H.; Ye, W. The role of epithelial-mesenchymal transition in regulating radioresistance. Crit. Rev. Oncol. Hematol. 2020, 150, 102961. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Cubillo, E.; Sarrió, D.; Peinado, H.; Rodríguez-Pinilla, S.M.; Villa, S.; Bolós, V.; Jordá, M.; Fabra, A.; Portillo, F.; et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9543–9556. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef]
- Menju, T.; Date, H. Lung cancer and epithelial-mesenchymal transition. Gen. Thorac. Cardiovasc. Surg. 2021, 69, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, K.; Li, Z.; Zhang, Y.; Mu, L.; Chen, M.; Wang, R.; Deng, W.; Zou, L.; Liu, J. β-Elemene enhances radiosensitivity in non-small-cell lung cancer by inhibiting epithelial-mesenchymal transition and cancer stem cell traits via Prx-1/NF-kB/iNOS signaling pathway. Aging 2020, 13, 2575–2592. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, H.J.; Jung, C.W.; Lee, T.S.; Kim, E.H.; Park, M.J. CXCR4 uses STAT3-mediated slug expression to maintain radioresistance of non-small cell lung cancer cells: Emerges as a potential prognostic biomarker for lung cancer. Cell Death Dis. 2021, 12, 48. [Google Scholar] [CrossRef]
- Shih, P.C.; Mei, K.C. Role of STAT3 signaling transduction pathways in cancer stem cell-associated chemoresistance. Drug Discov. Today 2021, 26, 1450–1458. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, S.I.; Kim, R.K.; Cho, E.W.; Kim, I.G. Tescalcin/c-Src/IGF1Rβ-mediated STAT3 activation enhances cancer stemness and radioresistant properties through ALDH1. Sci. Rep. 2018, 8, 10711. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.W.; Peng, P.; Zhao, W.H.; Tian, Y.J.; Huang, Y.; Xia, S.; Chen, Y. mTORC1 inhibitor RAD001 (everolimus) enhances non-small cell lung cancer cell radiosensitivity in vitro via suppressing epithelial-mesenchymal transition. Acta Pharm. Sin. 2019, 40, 1085–1094. [Google Scholar] [CrossRef]
- Cong, L.; Yi, J.; Qiu, S.; Wang, R.; Jin, S.; Jiang, R.; Cong, X. Effect of EG00229 on Radiation Resistance of Lung Adenocarcinoma Cells. J. Cancer 2021, 12, 6105–6117. [Google Scholar] [CrossRef]
- Lu, Y.; Ma, J.; Li, Y.; Huang, J.; Zhang, S.; Yin, Z.; Ren, J.; Huang, K.; Wu, G.; Yang, K.; et al. CDP138 silencing inhibits TGF-β/Smad signaling to impair radioresistance and metastasis via GDF15 in lung cancer. Cell Death Dis. 2017, 8, e3036. [Google Scholar] [CrossRef]
- Cui, Y.H.; Kang, J.H.; Suh, Y.; Zhao, Y.; Yi, J.M.; Bae, I.H.; Lee, H.J.; Park, D.W.; Kim, M.J.; Lee, S.J. Loss of FBXL14 promotes mesenchymal shift and radioresistance of non-small cell lung cancer by TWIST1 stabilization. Signal Transduct. Target. 2021, 6, 272. [Google Scholar] [CrossRef]
- Lu, J.; Zhan, Y.; Feng, J.; Luo, J.; Fan, S. MicroRNAs associated with therapy of non-small cell lung cancer. Int. J. Biol. Sci. 2018, 14, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.M.; Yu, H.; Yuan, Y.J.; Zhang, J.; Ma, Y.; Cao, X.C.; Wang, J.; Zhao, L.J.; Wang, P. Overcoming of Radioresistance in Non-small Cell Lung Cancer by microRNA-320a Through HIF1α-Suppression Mediated Methylation of PTEN. Front. Cell Dev. Biol. 2020, 8, 553733. [Google Scholar] [CrossRef] [PubMed]
- Arechaga-Ocampo, E.; Lopez-Camarillo, C.; Villegas-Sepulveda, N.; Gonzalez-De la Rosa, C.H.; Perez-Añorve, I.X.; Roldan-Perez, R.; Flores-Perez, A.; Peña-Curiel, O.; Angeles-Zaragoza, O.; Rangel Corona, R.; et al. Tumor suppressor miR-29c regulates radioresistance in lung cancer cells. Tumour Biol. 2017, 39, 1010428317695010. [Google Scholar] [CrossRef] [PubMed]
- Cellini, F.; Morganti, A.G.; Genovesi, D.; Silvestris, N.; Valentini, V. Role of microRNA in response to ionizing radiations: Evidences and potential impact on clinical practice for radiotherapy. Molecules 2014, 19, 5379–5401. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, S.; Chen, X.; Feng, G.; Chen, Z.; Fan, S. MiR-145 modulates the radiosensitivity of non-small cell lung cancer cells by suppression of TMOD3. Carcinogenesis 2022, 43, 288–296. [Google Scholar] [CrossRef]
- Wei, W.; Dong, Z.; Gao, H.; Zhang, Y.-Y.; Shao, L.-H.; Jin, L.-L.; Lv, Y.-H.; Zhao, G.; Shen, Y.-N.; Jin, S.-Z. MicroRNA-9 enhanced radiosensitivity and its mechanism of DNA methylation in non-small cell lung cancer. Gene 2019, 710, 178–185. [Google Scholar] [CrossRef]
- Ma, W.; Ma, C.N.; Zhou, N.N.; Li, X.D.; Zhang, Y.J. Up- regulation of miR-328-3p sensitizes non-small cell lung cancer to radiotherapy. Sci. Rep. 2016, 6, 31651. [Google Scholar] [CrossRef]
- Xue, T.; Yin, G.; Yang, W.; Chen, X.; Liu, C.; Yang, W.; Zhu, J. MiR-129-5p Promotes Radio-sensitivity of NSCLC Cells by Targeting SOX4 and RUNX1. Curr. Cancer Drug Targets 2021, 21, 702–712. [Google Scholar] [CrossRef]
- Chen, G.; Yu, L.; Dong, H.; Liu, Z.; Sun, Y. MiR-182 enhances radioresistance in non-small cell lung cancer cells by regulating FOXO3. Clin. Exp. Pharmacol. Physiol. 2019, 46, 137–143. [Google Scholar] [CrossRef]
- Yuan, Y.; Liao, H.; Pu, Q.; Ke, X.; Hu, X.; Ma, Y.; Luo, X.; Jiang, Q.; Gong, Y.; Wu, M.; et al. miR-410 induces both epithelial–mesenchymal transition and radioresistance through activation of the PI3K/mTOR pathway in non-small cell lung cancer. Signal Transduct. Target. Ther. 2020, 5, 85. [Google Scholar] [CrossRef]
- Li, L.; Zhu, T.; Gao, Y.F.; Zheng, W.; Wang, C.J.; Xiao, L.; Huang, M.S.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. Targeting DNA Damage Response in the Radio(Chemo)therapy of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 839. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Almasan, A. USP14 Regulates DNA Damage Response and Is a Target for Radiosensitization in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2020, 21, 6383. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.S. DNA Double-Strand Breaks: A Double-Edged Sword for Trypanosomatids. Front. Cell Dev. Biol. 2021, 9, 669041. [Google Scholar] [CrossRef]
- Rahimian, E.; Amini, A.; Alikarami, F.; Pezeshki, S.M.S.; Saki, N.; Safa, M. DNA repair pathways as guardians of the genome: Therapeutic potential and possible prognostic role in hematologic neoplasms. DNA Repair. 2020, 96, 102951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, Q.; Zhu, L.; Xie, S.; Tu, L.; Yang, Y.; Wu, K.; Zhao, Y.; Wang, Y.; Xu, Y.; et al. SERPINE2/PN-1 regulates the DNA damage response and radioresistance by activating ATM in lung cancer. Cancer Lett. 2022, 524, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Chen, Y.; Zhao, S.; Huang, Y.; Liu, T.; Liu, H.; Li, B.; Cui, J.; Cai, J.; Bai, C.; et al. Sirt3 Promoted DNA Damage Repair and Radioresistance Through ATM-Chk2 in Non-small Cell Lung Cancer Cells. J. Cancer 2021, 12, 5464–5472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, C.; Tan, Y.; Zhang, H.; Li, Y.; Zou, H. ITGB1 enhances the Radioresistance of human Non-small Cell Lung Cancer Cells by modulating the DNA damage response and YAP1-induced Epithelial-mesenchymal Transition. Int. J. Biol. Sci. 2021, 17, 635–650. [Google Scholar] [CrossRef]
- Kitabatake, K.; Yoshida, E.; Kaji, T.; Tsukimoto, M. Involvement of adenosine A2B receptor in radiation-induced translocation of epidermal growth factor receptor and DNA damage response leading to radioresistance in human lung cancer cells. Biochim. Et Biophys. Acta Gen. Subj. 2020, 1864, 129457. [Google Scholar] [CrossRef]
- Ai, X.; Guo, X.; Wang, J.; Stancu, A.L.; Joslin, P.M.N.; Zhang, D.; Zhu, S. Targeted therapies for advanced non-small cell lung cancer. Oncotarget 2018, 9, 37589–37607. [Google Scholar] [CrossRef]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef]
- Díaz-Serrano, A.; Gella, P.; Jiménez, E.; Zugazagoitia, J.; Paz-Ares Rodríguez, L. Targeting EGFR in Lung Cancer: Current Standards and Developments. Drugs 2018, 78, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Anaplastic lymphoma kinase (ALK) inhibitors in the treatment of ALK-driven lung cancers. Pharmacol. Res. 2017, 117, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Barbacid, M. Targeting KRAS mutant lung cancer: Light at the end of the tunnel. Mol. Oncol. 2022, 16, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-G.; Shih, J.-Y. Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Wagener-Ryczek, S.; Heydt, C.; Süptitz, J.; Michels, S.; Falk, M.; Alidousty, C.; Fassunke, J.; Ihle, M.A.; Tiemann, M.; Heukamp, L.; et al. Mutational spectrum of acquired resistance to reversible versus irreversible EGFR tyrosine kinase inhibitors. BMC Cancer 2020, 20, 408. [Google Scholar] [CrossRef]
- Lamb, Y.N. Osimertinib: A Review in Previously Untreated, EGFR Mutation-Positive, Advanced NSCLC. Target. Oncol. 2021, 16, 687–695. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M–Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, J.; Yan, H.; Cheng, Y.; Wang, Y.; Yang, Y.; Deng, M.; Che, X.; Hou, K.; Qu, X.; et al. Lymecycline reverses acquired EGFR-TKI resistance in non–small-cell lung cancer by targeting GRB2. Pharmacol. Res. 2020, 159, 105007. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, X.; Ren, Y.; Luo, J.; Liu, P.; Su, D.; Yang, X. Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway. Cell Death Dis. 2018, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Zheng, J.; Chen, M.; Jiang, N.; Xu, X.; Huang, F. HIF-1 Inhibitor YC-1 Reverses the Acquired Resistance of EGFR-Mutant HCC827 Cell Line with MET Amplification to Gefitinib. Oxid. Med. Cell. Longev. 2021, 2021, 6633867. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Miao, J.; Sun, R.; Wang, S.; Molina-Vila, M.A.; Chaib, I.; Rosell, R.; Cao, P. Dihydroartemisinin overcomes the resistance to osimertinib in EGFR-mutant non-small-cell lung cancer. Pharmacol. Res. 2021, 170, 105701. [Google Scholar] [CrossRef] [PubMed]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non–Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef]
- Dey, S.; Ashrafi, A.; Vidal, C.; Jain, N.; Kalainayakan, S.P.; Ghosh, P.; Alemi, P.S.; Salamat, N.; Konduri, P.C.; Kim, J.W.; et al. Heme Sequestration Effectively Suppresses the Development and Progression of Both Lung Adenocarcinoma and Squamous Cell Carcinoma. Mol. Cancer Res. 2022, 20, 139–149. [Google Scholar] [CrossRef]
- Wang, T.; Ashrafi, A.; Konduri, P.C.; Ghosh, P.; Dey, S.; Modareszadeh, P.; Salamat, N.; Alemi, P.S.; Berisha, E.; Zhang, L. Heme Sequestration as an Effective Strategy for the Suppression of Tumor Growth and Progression. Mol. Cancer Ther. 2021, 20, 2506–2518. [Google Scholar] [CrossRef]
- Elliott, J.; Bai, Z.; Hsieh, S.-C.; Kelly, S.E.; Chen, L.; Skidmore, B.; Yousef, S.; Zheng, C.; Stewart, D.J.; Wells, G.A. ALK inhibitors for non-small cell lung cancer: A systematic review and network meta-analysis. PLoS ONE 2020, 15, e0229179. [Google Scholar] [CrossRef]
- Camidge, D.R.; Dziadziuszko, R.; Peters, S.; Mok, T.; Noe, J.; Nowicka, M.; Gadgeel, S.M.; Cheema, P.; Pavlakis, N.; de Marinis, F.; et al. Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-Positive Advanced Non–Small Cell Lung Cancer in the Global Phase III ALEX Study. J. Thorac. Oncol. 2019, 14, 1233–1243. [Google Scholar] [CrossRef]
- Gainor, J.F.; Varghese, A.M.; Ou, S.H.; Kabraji, S.; Awad, M.M.; Katayama, R.; Pawlak, A.; Mino-Kenudson, M.; Yeap, B.Y.; Riely, G.J.; et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: An analysis of 1683 patients with non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Gristina, V.; La Mantia, M.; Iacono, F.; Galvano, A.; Russo, A.; Bazan, V. The Emerging Therapeutic Landscape of ALK Inhibitors in Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA Approval Summary: Crizotinib for the Treatment of Metastatic Non-Small Cell Lung Cancer with Anaplastic Lymphoma Kinase Rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, P.; Yuan, Y.; Deng, X.H.; Yue, R.; Ge, X.Z. Efficacy and safety of ceritinib in anaplastic lymphoma kinase-rearranged non-small cell lung cancer: A systematic review and meta-analysis. J. Clin. Pharm. 2020, 45, 743–754. [Google Scholar] [CrossRef]
- Paik, J.; Dhillon, S. Alectinib: A Review in Advanced, ALK-Positive NSCLC. Drugs 2018, 78, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Peng, L.; Lu, D.; Xia, Y.; Hong, S.; Selvaggi, G.; Stebbing, J.; Sun, Y.; Liang, F. Efficacy and Safety of First-Line Treatment Strategies for Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer: A Bayesian Network Meta-Analysis. Front. Oncol. 2021, 11, 754768. [Google Scholar] [CrossRef]
- Rothenstein, J.M.; Chooback, N. ALK Inhibitors, Resistance Development, Clinical Trials. Curr. Oncol. 2018, 25, 59–67. [Google Scholar] [CrossRef]
- Horn, L.; Wang, Z.; Wu, G.; Poddubskaya, E.; Mok, T.; Reck, M.; Wakelee, H.; Chiappori, A.A.; Lee, D.H.; Breder, V.; et al. Ensartinib vs Crizotinib for Patients With Anaplastic Lymphoma Kinase−Positive Non–Small Cell Lung Cancer: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Nagasaka, M.; Brazel, D.; Hou, Y.; Zhu, V.W. Will the clinical development of 4th-generation “double mutant active” ALK TKIs (TPX-0131 and NVL-655) change the future treatment paradigm of ALK+ NSCLC? Transl. Oncol. 2021, 14, 101191. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-generation EGFR and ALK inhibitors: Mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 2022, 19, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2021, 29, 875–878. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Rao, S.; Mondragón, L.; Pranjic, B.; Hanada, T.; Stoll, G.; Köcher, T.; Zhang, P.; Jais, A.; Lercher, A.; Bergthaler, A.; et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Res. 2019, 29, 579–591. [Google Scholar] [CrossRef]
- Rebane-Klemm, E.; Truu, L.; Reinsalu, L.; Puurand, M.; Shevchuk, I.; Chekulayev, V.; Timohhina, N.; Tepp, K.; Bogovskaja, J.; Afanasjev, V.; et al. Mitochondrial Respiration in KRAS and BRAF Mutated Colorectal Tumors and Polyps. Cancers 2020, 12, 815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Sukari, A.; Nagasaka, M.; Al-Hadidi, A.; Lum, L.G. Cancer Immunology and Immunotherapy. Anticancer Res. 2016, 36, 5593. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. Advances in cancer immunology and cancer immunotherapy. Discov. Med. 2016, 21, 125–133. [Google Scholar]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients With Advanced Non-Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef]
- Bodor, J.N.; Kasireddy, V.; Borghaei, H. First-Line Therapies for Metastatic Lung Adenocarcinoma Without a Driver Mutation. J. Oncol. Pract. 2018, 14, 529–535. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Rath, B. Immunotherapy for small cell lung cancer: Mechanisms of resistance. Expert Opin. Biol. 2019, 19, 423–432. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res. 2020, 8, 34. [Google Scholar] [CrossRef]
- Gkountakos, A.; Delfino, P.; Lawlor, R.T.; Scarpa, A.; Corbo, V.; Bria, E. Harnessing the epigenome to boost immunotherapy response in non-small cell lung cancer patients. Adv. Med. Oncol. 2021, 13, 17588359211006947. [Google Scholar] [CrossRef]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de Los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to Durvalumab and Durvalumab plus Tremelimumab Is Associated with Functional STK11 Mutations in Patients with Non-Small Cell Lung Cancer and Is Reversed by STAT3 Knockdown. Cancer Discov. 2021, 11, 2828–2845. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Gutiontov, S.I.; Turchan, W.T.; Spurr, L.F.; Rouhani, S.J.; Chervin, C.S.; Steinhardt, G.; Lager, A.M.; Wanjari, P.; Malik, R.; Connell, P.P.; et al. CDKN2A loss-of-function predicts immunotherapy resistance in non-small cell lung cancer. Sci. Rep. 2021, 11, 20059. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Yu, Y.; Lan, F.; Yan, J.; Li, J.; Li, W.; Xia, Y. Case Report: Tumor Microenvironment Characteristics in a Patient With HER2 Mutant Lung Squamous Cell Carcinoma Harboring High PD-L1 Expression Who Presented Hyperprogressive Disease. Front. Oncol. 2021, 11, 760703. [Google Scholar] [CrossRef] [PubMed]
- Horvath, L.; Thienpont, B.; Zhao, L.; Wolf, D.; Pircher, A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)—Novel approaches and future outlook. Mol. Cancer 2020, 19, 141. [Google Scholar] [CrossRef]
- Ren, S.; Xiong, X.; You, H.; Shen, J.; Zhou, P. The Combination of Immune Checkpoint Blockade and Angiogenesis Inhibitors in the Treatment of Advanced Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 689132. [Google Scholar] [CrossRef]
- Huang, M.; Xiong, D.; Pan, J.; Zhang, Q.; Wang, Y.; Myers, C.R.; Johnson, B.D.; Hardy, M.; Kalyanaraman, B.; You, M. Prevention of Tumor Growth and Dissemination by In Situ Vaccination with Mitochondria-Targeted Atovaquone. Adv. Sci. 2022, 9, e2101267. [Google Scholar] [CrossRef]
- Kopecka, J.; Salaroglio, I.C.; Perez-Ruiz, E.; Sarmento-Ribeiro, A.B.; Saponara, S.; De Las Rivas, J.; Riganti, C. Hypoxia as a driver of resistance to immunotherapy. Drug Resist. Updat 2021, 59, 100787. [Google Scholar] [CrossRef]
- Boreel, D.F.; Span, P.N.; Heskamp, S.; Adema, G.J.; Bussink, J. Targeting Oxidative Phosphorylation to Increase the Efficacy of Radio- and Immune-Combination Therapy. Clin. Cancer Res. 2021, 27, 2970–2978. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, M.X.; Li, K.S.; Peng, L.; Zhang, P.F. Circular RNA drives resistance to anti-PD-1 immunotherapy by regulating the miR-30a-5p/SOX4 axis in non-small cell lung cancer. Cancer Drug Resist. 2022, 5, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.H.; Yang, Y.P.; Chien, C.S.; Yarmishyn, A.A.; Adekunle Ishola, A.; Chien, Y.; Chen, Y.M.; Tsai, P.H.; Lin, T.W.; Wang, M.L.; et al. Circular RNA hsa_circ_0000190 Facilitates the Tumorigenesis and Immune Evasion by Upregulating the Expression of Soluble PD-L1 in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 23, 64. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, T.; She, Y.; Wu, K.; Gu, S.; Li, L.; Dong, C.; Chen, C.; Zhou, Y. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol. Cancer 2021, 20, 105. [Google Scholar] [CrossRef]
- Zhang, P.F.; Pei, X.; Li, K.S.; Jin, L.N.; Wang, F.; Wu, J.; Zhang, X.M. Circular RNA circFGFR1 promotes progression and anti-PD-1 resistance by sponging miR-381-3p in non-small cell lung cancer cells. Mol. Cancer 2019, 18, 179. [Google Scholar] [CrossRef]
- Chen, S.W.; Zhu, S.Q.; Pei, X.; Qiu, B.Q.; Xiong, D.; Long, X.; Lin, K.; Lu, F.; Xu, J.J.; Wu, Y.B. Cancer cell-derived exosomal circUSP7 induces CD8(+) T cell dysfunction and anti-PD1 resistance by regulating the miR-934/SHP2 axis in NSCLC. Mol. Cancer 2021, 20, 144. [Google Scholar] [CrossRef]
- Huang, J.; Liu, D.; Wang, Y.; Liu, L.; Li, J.; Yuan, J.; Jiang, Z.; Jiang, Z.; Hsiao, W.W.; Liu, H.; et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumour effect of antiprogrammed cell death 1/programmed cell death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut 2022, 71, 734–745. [Google Scholar] [CrossRef]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2020, 25, e147–e159. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Hernández, A.; Del Barco Morillo, E.; Parra Pérez, C.; Miramontes-González, J.P.; Figuero-Pérez, L.; Martín-Gómez, T.; Escala-Cornejo, R.; Bellido Hernández, L.; González Sarmiento, R.; Cruz-Hernández, J.J.; et al. Influence of DNA Mismatch Repair (MMR) System in Survival and Response to Immune Checkpoint Inhibitors (ICIs) in Non-Small Cell Lung Cancer (NSCLC): Retrospective Analysis. Biomedicines 2022, 10, 360. [Google Scholar] [CrossRef]
- Aggarwal, C.; Somaiah, N.; Simon, G. Antiangiogenic agents in the management of non-small cell lung cancer: Where do we stand now and where are we headed? Cancer Biol. 2012, 13, 247–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the immune response by pro-angiogenic factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.; Jänne, P.; Opyrchal, M.; Hafez, N.; Raez, L.; Gabrilovich, D.; Wang, F.; Ordentlich, P.; Brouwer, S.; Sankoh, S.; et al. OA05.01 Efficacy/Safety of Entinostat (ENT) and Pembrolizumab (PEMBRO) in NSCLC Patients Previously Treated with Anti-PD-(L)1 Therapy. J. Thorac. Oncol. 2018, 13, S330. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- He, D.; Li, X.; An, R.; Wang, L.; Wang, Y.; Zheng, S.; Chen, X.; Wang, X. Response to PD-1-Based Immunotherapy for Non-Small Cell Lung Cancer Altered by Gut Microbiota. Oncol. Ther. 2021, 9, 647–657. [Google Scholar] [CrossRef]
- Fang, C.; Fang, W.; Xu, L.; Gao, F.; Hou, Y.; Zou, H.; Ma, Y.; Moll, J.M.; Yang, Y.; Wang, D.; et al. Distinct Functional Metagenomic Markers Predict the Responsiveness to Anti-PD-1 Therapy in Chinese Non-Small Cell Lung Cancer Patients. Front. Oncol. 2022, 12, 837525. [Google Scholar] [CrossRef]
- Insinga, R.P.; Feliciano, J.L.; Qiao, N.; Vandormael, K.; Zhang, Y. Cost-effectiveness of pembrolizumab + chemotherapy versus chemotherapy and pembrolizumab monotherapy in first line treatment of NSCLC in the US—Updated analyses with additional trial follow-up. J. Med. Econ. 2021, 24, 792–805. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-Y.; Jiang, X.-M.; Wang, B.-L.; Sun, Y.; Lu, J.-J. Combination therapy with PD-1/PD-L1 blockade in non-small cell lung cancer: Strategies and mechanisms. Pharmacol. Ther. 2021, 219, 107694. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.; Rodríguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Dómine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Reck, M.; Ciuleanu, T.-E.; Dols, M.C.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. J. Clin. Oncol. 2020, 38, 9501. [Google Scholar] [CrossRef]
- Galffy, G.; Lugowska, I.; Poddubskaya, E.; Cho, B.C.; Ahn, M.-J.; Han, J.-Y.; Su, W.-C.; Hauke, R.; Dyar, S.; Lee, D.H.; et al. 281 JAVELIN Medley VEGF: Phase 2 study of avelumab + axitinib in patients with previously treated non-small cell lung cancer (NSCLC) or treatment naive, cisplatin-ineligible urothelial cancer (UC). J. ImmunoTherapy Cancer 2020, 8, A171. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Theelen, W.S.M.E.; Peulen, H.M.U.; Lalezari, F.; van der Noort, V.; de Vries, J.F.; Aerts, J.G.J.V.; Dumoulin, D.W.; Bahce, I.; Niemeijer, A.-L.N.; de Langen, A.J.; et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non–Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1276–1282. [Google Scholar] [CrossRef]
- Morganti, S.; Curigliano, G. Combinations using checkpoint blockade to overcome resistance. Ecancermedicalscience 2020, 14, 1148. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Tricker, E.M.; Xu, C.; Uddin, S.; Capelletti, M.; Ercan, D.; Ogino, A.; Pratilas, C.A.; Rosen, N.; Gray, N.S.; Wong, K.-K.; et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 2015, 5, 960–971. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Jiao, D.; Yang, S. Overcoming Resistance to Drugs Targeting KRAS Mutation. Innovation 2020, 1, 100035. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharm. 2018, 9, 185. [Google Scholar] [CrossRef]
- Schoenhals, J.E.; Seyedin, S.N.; Tang, C.; Cortez, M.A.; Niknam, S.; Tsouko, E.; Chang, J.Y.; Hahn, S.M.; Welsh, J.W. Preclinical Rationale and Clinical Considerations for Radiotherapy Plus Immunotherapy: Going Beyond Local Control. Cancer J. 2016, 22, 130–137. [Google Scholar] [CrossRef]
- Gong, X.; Li, X.; Jiang, T.; Xie, H.; Zhu, Z.; Zhou, F.; Zhou, C. Combined Radiotherapy and Anti-PD-L1 Antibody Synergistically Enhances Antitumor Effect in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1085–1097. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Alfaro, C.; Sanchez, J.C.; Franco, F.; Calvo, V.; et al. Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-β signaling. Free Radic. Biol. Med. 2019, 130, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.M.; Wingert, R.A. PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef] [PubMed]
- Paredes, F.; Sheldon, K.; Lassègue, B.; Williams, H.C.; Faidley, E.A.; Benavides, G.A.; Torres, G.; Sanhueza-Olivares, F.; Yeligar, S.M.; Griendling, K.K.; et al. Poldip2 is an oxygen-sensitive protein that controls PDH and αKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 1789–1794. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat Res. 2016, 57 (Suppl. S1), i90–i98. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mourón, S.; Muñoz, J.; Gómez-López, G.; Jimenez-Renard, V.; Mulero, F.; et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Ghosh, P.; Guo, Y.; Ashrafi, A.; Chen, J.; Dey, S.; Zhong, S.; Liu, J.; Campbell, J.; Konduri, P.C.; Gerberich, J.; et al. Oxygen-Enhanced Optoacoustic Tomography Reveals the Effectiveness of Targeting Heme and Oxidative Phosphorylation at Normalizing Tumor Vascular Oxygenation. Cancer Res. 2020, 80, 3542–3555. [Google Scholar] [CrossRef]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Livney, Y.D.; Assaraf, Y.G. Rationally designed nanovehicles to overcome cancer chemoresistance. Adv. Drug Deliv. Rev. 2013, 65, 1716–1730. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mode of Action | Target Entity | Chemotherapeutic Agent | References |
|---|---|---|---|
| DNA repair system | Upregulation of: ERCC1 DNA Polymerase | Platinum compounds | [94,95] |
| Drug efflux | Upregulation of ABC family transporters: ABCC1/MRP1 ABCC3/MRP3 ABCB1/MDR1 ABCC10/MRP7 ABCB1/MDR1/p-glycoprotein ABCC6/MRP6 ABCC11/MRP8 | Platinum compounds Microtubule-targeted compounds Etoposide Gemcitabine Pemetrexed | [33,34,35,36,37,38] |
| Prosurvival signaling | Upregulation of: EGFR PI3K/Akt MAPK Calpain Sphk1 | Platinum compounds Microtubule-targeted compounds Pemetrexed | [42,43,44] |
| Cell cycle arrest | Upregulation of: Bcl-2 Bcl-XL Autophagy | Platinum compounds Gemcitabine Microtubule-targeted compounds | [46,47,96] |
| Epigenetic regulation | Promoter methylation of IGFBP3 and FOXF1 Upregulation of KDM3B Deregulation of circadian rhythm | Platinum compounds | [67,97,98,99] |
| MicroRNA | Upregulation of: miR-106a, miR- 31, miR-15b, miR-27a, miR-223, miR-205, miR-92b, miR-224, miR-34c-5p, miR-181a, miR-135a, miR 197-3p, miR-222-3p downregulation of: miR-101-3p, miR-181, miR-589, miR-1244, miR-29c, miR-630, and miR-197 miR-16, miR-17-5p, miR-216b, miR-200b, miR-363-3p | Platinum compounds Microtubule-targeted compounds Etoposide Gemcitabine | [16,84,85,86,87] |
| EMT/CSC | Upregulation of: EMT phenotype Notch signaling Wnt signaling Shh signaling | Platinum compounds Microtubule-targeted compounds Etoposide Pemetrexed | [100,101] |
| Tumor microenvironment | Upregulation of: Hypoxia CAF PDL-1 | Platinum compounds | [102,103,104] |
| Cancer metabolism | Upregulation of: PGC1α and glutamine metabolism Downregulation of: OXPHOS and glycolysis | Platinum compounds | [90,105] |
| Resistance Mode | Action | Target | References |
|---|---|---|---|
| Gene mutations | a. Modify the expression of PD-1, PD-L1, and CTLA-4 proteins b. Deregulate PD-1/PD-L1 signaling pathways | ALK, EGFR, HER-2, CDK2NA, STK11 (LKB1); TP53; KRAS | [219,221,226,227] |
| Dysregulation of cellular and molecular pathways | a. Promote primary and adaptive resistance to anti-CTLA-4 and anti-PD-1 b. Enhance cancer cell proliferation and metastasis | Wnt/B-catenin, JAK/STAT3, PI3K-Akt, JAK1/2 mutations, IFN-γ signaling pathways | [215,222,224] |
| Neo-angiogenesis | a. Inhibit the infiltration of effector immune cells b. Upregulate the expression of PD-L1 c. Recruit Treg, TAM, and MDSC cells d. Impair the delivery of therapeutic agents to tumor cells e. Reduce adhesion molecules into TME | Hypoxia, HIF1-A, VEGFA, VEGFR, Angiopoietin-2 (ANG2) | [228,229] |
| High oxidative metabolism | a. Trigger hypoxia b. Promote cancer cell growth, proliferation, and metastasis c. Create immunosuppressive TME d. Affect effector T cells | OXPHOS complexes, heme, HIF1-A, VEGFA, VEGFR | [179,180,230,231,232] |
| Immunosuppressive TME | a. Recruit immunosuppressive cells (Tregs, Bregs, MDSCs, TAM, and CAF) b. Inhibit the infiltration of the effector immune cells c. Release of proinflammatory molecules d. Upregulate immune checkpoint proteins (ICPs) e. Affect antitumor immunity | Alternative ICPs, immunosuppressive molecules, proinflammatory molecules, VEGFA, HIF1-A | [228,233,234] |
| Upregulation of alternative immune checkpoints | a. Modulate TME and show adaptive resistance to anti-PD-1 | LAG-3, TIGIT, TIM3, and TIM-1 | [228,234] |
| Deregulated epigenetics | a. Modify the expression of immune related genes b. Triggers T-cell dysfunction | DNA methyl transferase; histone methyl transferase; histone deacetylase | [215,218] |
| Dysregulated circRNAs | a. Upregulate PD-L1 b. Recruit inflammatory molecules c. Inhibit the CD8+ T cells’ infiltration into tumorigenic regions | hsa_circ_0000190, hsa_circ_0079587, circFGFR1, circUSP7 | [235,236,237,238,239] |
| Modified gut microbiota | a. Reduce effector T cells b. Increase regulatory T cells c. Affect antitumor responses | Gut microbiota composition; gut bacteria | [240] |
| Drug Combination | Phases/Study | Treatment Outcome | References |
|---|---|---|---|
| Platinum + pemetrexed + pembrolizumab | Phase III (Keynote 189) | median OS—22.0 months; median PFS—9.0 months | [256] |
| Carboplatin + (nab)-paclitaxel + pembrolizumab | Phase III (Keynote 407) | median OS—15.9 months; median PFS—6.4 month | [257] |
| Carboplatin + nab-paclitaxel + atezolizumab | Phase III (Impower 130) | median OS—18.6 months; median PFS—7.0 months | [258] |
| Carboplatin + paclitaxel + bevacizumab + atezolizumab | Phase III (Impower 150) | median OS—19.2; median PFS—8.3 months | [253] |
| Pembrolizumab + platinum + pemetrexed | Phase III | median OS—12 months; median PFS—8.8 months | [255] |
| Nivolumab + ipilimumab + two cycles of chemotherapy | Phase III (CheckMate 9LA) | median OS—15.6 months; | [259] |
| Avelumab + axitinib | Phase II (Javelin Medley VEGF) | ORR—31.7%; median PFS—5.5 months | [260] |
| Nivolumab + ipilimumab | Phase III (CheckMate 227) | median OS—17.1 months | [261] |
| Pembrolizumab + stereotactic body radiation therapy (SBRT) | Phase II (PEMBRO-RT) | median OS—15.9 months; median PFS—6.6 months | [262] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Chacon Castro, M.d.C.; Deese, A.R.; Zhang, L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers 2022, 14, 4562. https://doi.org/10.3390/cancers14194562
Ashrafi A, Akter Z, Modareszadeh P, Modareszadeh P, Berisha E, Alemi PS, Chacon Castro MdC, Deese AR, Zhang L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers. 2022; 14(19):4562. https://doi.org/10.3390/cancers14194562
Chicago/Turabian StyleAshrafi, Adnin, Zakia Akter, Pouya Modareszadeh, Parsa Modareszadeh, Eranda Berisha, Parinaz Sadat Alemi, Maria del Carmen Chacon Castro, Alexander R. Deese, and Li Zhang. 2022. "Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance" Cancers 14, no. 19: 4562. https://doi.org/10.3390/cancers14194562
APA StyleAshrafi, A., Akter, Z., Modareszadeh, P., Modareszadeh, P., Berisha, E., Alemi, P. S., Chacon Castro, M. d. C., Deese, A. R., & Zhang, L. (2022). Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers, 14(19), 4562. https://doi.org/10.3390/cancers14194562