

Investigation of the Role of PUFA Metabolism in Breast Cancer Using a Rank-Based Random Forest Algorithm

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Random Forest Model
- From input data N × M (where N is the number of samples and M is the number of used features), k subsets are randomly selected with a return;
- A decision tree is built for each subset;
- The final decision is made on the majority vote for classification tasks or by averaging in the regression tasks.
2.3. Boruta Feature Selection Algorithm
2.4. Sequential Feature Selector for Minimal Gene Set Selection
2.5. SHAP Values to Identify the Most Important PUFA Genes
2.6. Enrichment Analysis
2.7. Differential Expression Analysis
3. Results
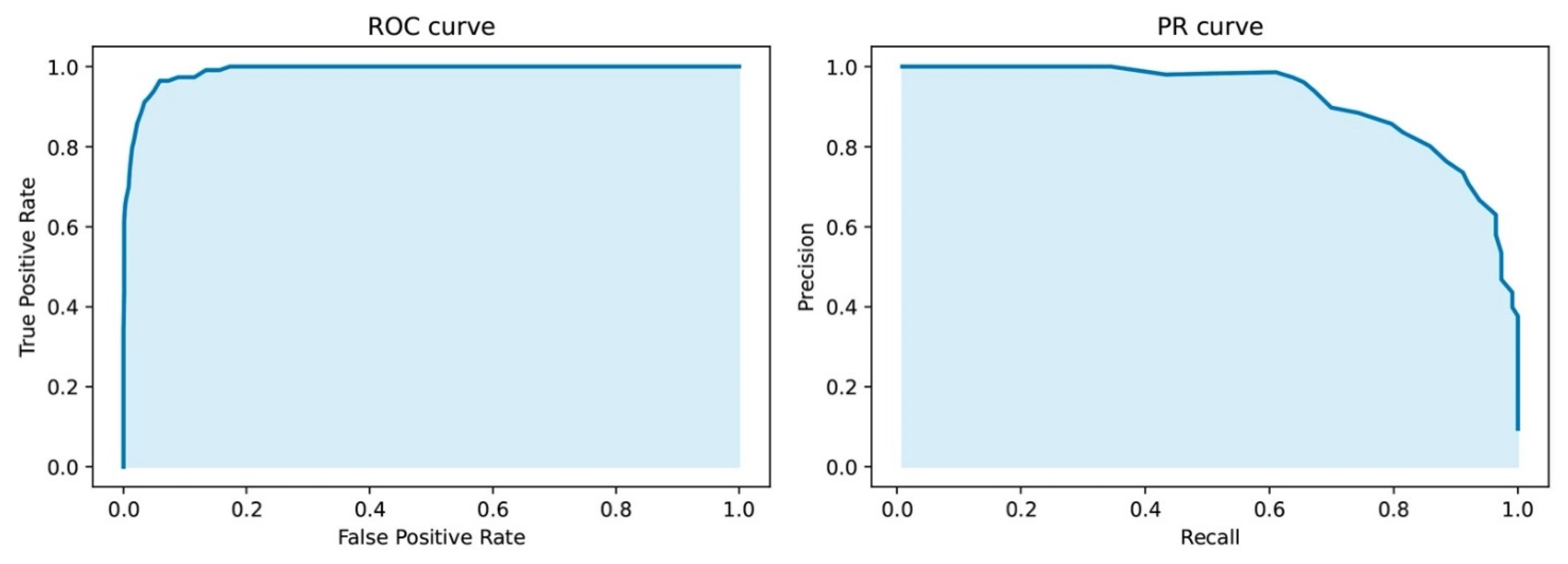
3.1. Validation of Machine Learning Nonparametric Approach
3.2. Rank Model to Identify Most Important PUFA Genes for Breast Cancer vs. Normal Tissues Classification
3.3. Rank Model to Identify Most Important PUFA Genes for Breast Cancer Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Kensler, K.H.; Sankar, V.N.; Wang, J.; Zhang, X.; Rubadue, C.A.; Baker, G.M.; Parker, J.S.; Hoadley, K.A.; Stancu, A.L.; Pyle, M.E.; et al. PAM50 molecular intrinsic subtypes in the nurses’ health Study cohorts. Cancer Epidemiol. Biomarkers Prev. 2019, 28, 798–806. [Google Scholar] [CrossRef]
- Yu, T.J.; Ma, D.; Liu, Y.Y.; Xiao, Y.; Gong, Y.; Jiang, Y.Z.; Shao, Z.M.; Hu, X.; Di, G.H. Bulk and single-cell transcriptome profiling reveal the metabolic heterogeneity in human breast cancers. Mol. Ther. 2021, 29, 2350–2365. [Google Scholar] [CrossRef]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Wang, Y.P.; Li, J.T.; Qu, J.; Yin, M.; Lei, Q.Y. Metabolite sensing and signaling in cancer. J. Biol. Chem. 2020, 295, 11938–11946. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Sampath, H.; Ntambi, J.M. Polyunsaturated fatty acid regulation of gene expression. Nutr. Rev. 2004, 62, 727–739. [Google Scholar] [CrossRef]
- Jabbour, H.N.; Sales, K.J. Prostaglandin receptor signalling and function in human endometrial pathology. Trends Endocrinol. Metab. 2004, 15, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef]
- Schmid, T.; Brüne, B. Prostanoids and Resolution of Inflammation—Beyond the Lipid-Mediator Class Switch. Front. Immunol. 2021, 12, 714042. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Astakhova, A.A.; Sergeeva, M.G. Resolution of inflammation and mood disorders. Exp. Mol. Pathol. 2018, 105, 190–201. [Google Scholar] [CrossRef]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Johnson, A.M.; Kleczko, E.K.; Nemenoff, R.A. Eicosanoids in Cancer: New Roles in Immunoregulation. Front. Pharmacol. 2020, 11, 595498. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Role of prostanoids in gastrointestinal cancer. J. Clin. Investig. 2018, 128, 2732–2742. [Google Scholar] [CrossRef]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Sacco, A.; Sostres, C.; Bruno, A.; Dovizio, M.; Piazuelo, E.; Di Francesco, L.; Contursi, A.; Zucchelli, M.; Schiavone, S.; et al. Low-Dose Aspirin Acetylates Cyclooxygenase-1 in Human Colorectal Mucosa: Implications for the Chemoprevention of Colorectal Cancer. Clin. Pharmacol. Ther. 2017, 102, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Ma, X.; Kochel, T.; Goloubeva, O.; Staats, P.; Thompson, K.; Martin, S.; Reader, J.; Take, Y.; Collin, P.; et al. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res. Treat. 2014, 143, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Chen, E.P.; Smyth, E.M. Targeting COX-2 abrogates mammary tumorigenesis: Breaking cancer-associated suppression of immunosurveillance. Oncoimmunology 2014, 3, e29287. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef]
- Hanaka, H.; Pawelzik, S.C.; Johnsen, J.I.; Rakonjac, M.; Terawaki, K.; Rasmuson, A.; Sveinbjörnsson, B.; Schumacher, M.C.; Hamberg, M.; Samuelsson, B.; et al. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18757–18762. [Google Scholar] [CrossRef]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef]
- Chistyakov, D.V.; Grabeklis, S.; Goriainov, S.V.; Chistyakov, V.V.; Sergeeva, M.G.; Reiser, G. Astrocytes synthesize primary and cyclopentenone prostaglandins that are negative regulators of their proliferation. Biochem. Biophys. Res. Commun. 2018, 500, 204–210. [Google Scholar] [CrossRef]
- Wolf, I.; O’Kelly, J.; Rubinek, T.; Tong, M.; Nguyen, A.; Lin, B.T.; Tai, H.H.; Karlan, B.Y.; Koeffler, H.P. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006, 66, 7818–7823. [Google Scholar] [CrossRef]
- Kang, Y.P.; Yoon, J.H.; Long, N.P.; Koo, G.B.; Noh, H.J.; Oh, S.J.; Lee, S.B.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; et al. Spheroid-induced epithelial-mesenchymal transition provokes global alterations of breast cancer lipidome: A multi-layered omics analysis. Front. Oncol. 2019, 9, 145. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Pacheco-Velázquez, S.C.; Belmont-Díaz, J.A.; Robledo-Cadena, D.X.; Vargas-Navarro, J.L.; de la Peña, N.A.C.; Saavedra, E.; Moreno-Sánchez, R. Transcriptional Regulation of Energy Metabolism in Cancer Cells. Cells 2019, 8, 1225. [Google Scholar] [CrossRef] [PubMed]
- Soga, T. Cancer metabolism: Key players in metabolic reprogramming. Cancer Sci. 2013, 104, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The challenges of big data. Dis. Model. Mech. 2016, 9, 483–485. [Google Scholar] [CrossRef]
- Schmidt, B.; Hildebrandt, A. Next-generation sequencing: Big data meets high performance computing. Drug Discov. Today 2017, 22, 712–717. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Hossain, M.A.; Saiful Islam, S.M.; Quinn, J.M.W.; Huq, F.; Moni, M.A. Machine learning and bioinformatics models to identify gene expression patterns of ovarian cancer associated with disease progression and mortality. J. Biomed. Inform. 2019, 100, 103313. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef]
- Geman, D.; D’Avignon, C.; Naiman, D.Q.; Winslow, R.L. Classifying gene expression profiles from pairwise mRNA comparisons. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–19. [Google Scholar] [CrossRef]
- Tan, A.C.; Naiman, D.Q.; Xu, L.; Winslow, R.L.; Geman, D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics 2005, 21, 3896–3904. [Google Scholar] [CrossRef]
- Acharjee, A.; Larkman, J.; Xu, Y.; Cardoso, V.R.; Gkoutos, G.V. A random forest based biomarker discovery and power analysis framework for diagnostics research. BMC Med. Genomics 2020, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fortino, V.; Kinaret, P.; Fyhrquist, N.; Alenius, H.; Greco, D. A robust and accurate method for feature selection and prioritization from multi-class OMICs data. PLoS ONE 2014, 9, e107801. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, C.; Zhang, Y.; Liu, J.; Yu, B.; Liu, X.; Dehmer, M. Feature selection of gene expression data for Cancer classification using double RBF-kernels. BMC Bioinform. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Källberg, D.; Vidman, L.; Rydén, P. Comparison of Methods for Feature Selection in Clustering of High-Dimensional RNA-Sequencing Data to Identify Cancer Subtypes. Front. Genet. 2021, 12, 632620. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Raschka, S. MLxtend: Providing machine learning and data science utilities and extensions to Python’s scientific computing stack. J. Open Source Softw. 2018, 3, 638. [Google Scholar] [CrossRef]
- SequentialFeatureSelector: The Popular Forward and Backward Feature Selection Approaches Incl. Floating Variants—Mlxtend. Available online: http://rasbt.github.io/mlxtend/user_guide/feature_selection/SequentialFeatureSelector/ (accessed on 29 June 2022).
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Tejera, N.; Boeglin, W.E.; Suzuki, T.; Schneider, C. COX-2-dependent and -independent biosynthesis of dihydroxy-arachidonic acids in activated human leukocytes. J. Lipid Res. 2012, 53, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hajeyah, A.A.; Griffiths, W.J.; Wang, Y.; Finch, A.J.; O’Donnell, V.B. The Biosynthesis of Enzymatically Oxidized Lipids. Front. Endocrinol. 2020, 11, 591819. [Google Scholar] [CrossRef] [PubMed]
- Bryk, M.; Chwastek, J.; Kostrzewa, M.; Mlost, J.; Pędracka, A.; Starowicz, K. Alterations in anandamide synthesis and degradation during osteoarthritis progression in an animal model. Int. J. Mol. Sci. 2020, 21, 7381. [Google Scholar] [CrossRef] [PubMed]
- Chistyakov, D.V.; Guryleva, M.V.; Stepanova, E.S.; Makarenkova, L.M.; Ptitsyna, E.V.; Goriainov, S.V.; Nikolskaya, A.I.; Astakhova, A.A.; Klimenko, A.S.; Bezborodova, O.A.; et al. Multi-Omics Approach Points to the Importance of Oxylipins Metabolism in Early-Stage Breast Cancer. Cancers 2022, 14, 2041. [Google Scholar] [CrossRef] [PubMed]
- Gavrish, G.E.; Chistyakov, D.V.; Sergeeva, M.G. ARGEOS: A new bioinformatic tool for detailed systematics search in GEO and arrayexpress. Biology 2021, 10, 1026. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2010, 220, 263–280. [Google Scholar] [CrossRef]
- Pan, X.; Hu, X.H.; Zhang, Y.H.; Chen, L.; Zhu, L.C.; Wan, S.B.; Huang, T.; Cai, Y.D. Identification of the copy number variant biomarkers for breast cancer subtypes. Mol. Genet. Genomics 2019, 294, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Mohamed, E.M.; Xu, G.G.; Waters, M.; Jing, K.; Ma, Y.; Zhang, Y.; Spiegel, S.; Idowu, M.O.; Fang, X. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016, 7, 3832–3846. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Liu, J.; Jiang, W.Q.; Carew, J.S.; Ogasawara, M.A.; Pelicano, H.; Croce, C.M.; Estrov, Z.; Xu, R.H.; Keating, M.J.; et al. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene 2016, 35, 5663–5673. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Tomida, S.; Goodenowe, D.B.; Koyama, T.; Ozaki, E.; Kuriyama, N.; Morita, M.; Yamazaki, Y.; Sakaguchi, K.; Uehara, R.; Taguchi, T. Plasmalogen deficiency and overactive fatty acid elongation biomarkers in serum of breast cancer patients pre-and post-surgery—new insights on diagnosis, risk assessment, and disease mechanisms. Cancers 2021, 13, 4170. [Google Scholar] [CrossRef]
- Hunt, M.C.; Alexson, S.E.H. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog. Lipid Res. 2002, 41, 99–130. [Google Scholar] [CrossRef]
- Wang, F.; Wu, J.; Qiu, Z.; Ge, X.; Liu, X.; Zhang, C.; Xu, W.; Wang, F.; Hua, D.; Qi, X.; et al. ACOT1 expression is associated with poor prognosis in gastric adenocarcinoma. Hum. Pathol. 2018, 77, 35–44. [Google Scholar] [CrossRef]
- Jung, S.H.; Lee, H.C.; Hwang, H.J.; Park, H.A.; Moon, Y.A.; Kim, B.C.; Lee, H.M.; Kim, K.P.; Kim, Y.N.; Lee, B.L.; et al. Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCζ-p53-p21 signaling pathway. Cell Death Dis. 2017, 8, e2793. [Google Scholar] [CrossRef]
- Senga, S.; Kobayashi, N.; Kawaguchi, K.; Ando, A.; Fujii, H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-κB) signaling in cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1057–1067. [Google Scholar] [CrossRef]
- Cordero, A.; Kanojia, D.; Miska, J.; Panek, W.K.; Xiao, A.; Han, Y.; Bonamici, N.; Zhou, W.; Xiao, T.; Wu, M.; et al. FABP7 is a key metabolic regulator in HER2+ breast cancer brain metastasis. Oncogene 2019, 38, 6445–6460. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Zhang, Y.; Zhu, T.; Hua, Y.; Li, H.; Zhang, Q.; Xia, M. FABP4 promotes invasion and metastasis of colon cancer by regulating fatty acid transport. Cancer Cell Int. 2020, 20, 512. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Sauter, E.R.; Li, B. FABP4: A New Player in Obesity-Associated Breast Cancer. Trends Mol. Med. 2020, 26, 437–440. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Luminal A | Luminal B | HER2+ | Basal-Like |
|---|---|---|---|
| ELOVL5 | PTGES3 | FASN * | AKR1B1 |
| ACAA1 * | ADIPOR1 | FABP6 | CYP39A1 |
| PLD2 | MBOAT7 * | MGLL | PLD1 |
| ACAD8 * | ACOT8 * | ALOX15B | PLA2G4A |
| PLCL1 | CYP2B6 | FADS2 | FPR2 |
| HPGDS | FAAH | PLCG2 | |
| CYP4F11 | CYP7B1 | ||
| PTGER3 | FABP5 | ||
| CYP4F8 | PLA2G7 | ||
| ELOVL2 | CBR1 | ||
| EPHX2 | PLAA | ||
| LPCAT3 * | ACOT9 * | ||
| LTC4S | HSD17B12 | ||
| FABP4 | CYP39A1 | ||
| PLA2G2D | |||
| PLCH1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guryleva, M.V.; Penzar, D.D.; Chistyakov, D.V.; Mironov, A.A.; Favorov, A.V.; Sergeeva, M.G. Investigation of the Role of PUFA Metabolism in Breast Cancer Using a Rank-Based Random Forest Algorithm. Cancers 2022, 14, 4663. https://doi.org/10.3390/cancers14194663
Guryleva MV, Penzar DD, Chistyakov DV, Mironov AA, Favorov AV, Sergeeva MG. Investigation of the Role of PUFA Metabolism in Breast Cancer Using a Rank-Based Random Forest Algorithm. Cancers. 2022; 14(19):4663. https://doi.org/10.3390/cancers14194663
Chicago/Turabian StyleGuryleva, Mariia V., Dmitry D. Penzar, Dmitry V. Chistyakov, Andrey A. Mironov, Alexander V. Favorov, and Marina G. Sergeeva. 2022. "Investigation of the Role of PUFA Metabolism in Breast Cancer Using a Rank-Based Random Forest Algorithm" Cancers 14, no. 19: 4663. https://doi.org/10.3390/cancers14194663
APA StyleGuryleva, M. V., Penzar, D. D., Chistyakov, D. V., Mironov, A. A., Favorov, A. V., & Sergeeva, M. G. (2022). Investigation of the Role of PUFA Metabolism in Breast Cancer Using a Rank-Based Random Forest Algorithm. Cancers, 14(19), 4663. https://doi.org/10.3390/cancers14194663