

The Spliceosome: A New Therapeutic Target in Chronic Myeloid Leukaemia

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Primary Human Cell Samples
2.4. Reagent
2.5. Cell Viability Assays
2.6. Cell Counting, Apoptosis Assays and Cell Cycle Analysis
2.7. Cell Division Monitoring by Staining with Carboxyfluorescein Diacetate Succinimidyl Diester
2.8. Colony Forming Cell (CFC) Assays
2.9. Gene Expression
2.10. Statistical Analysis
3. Results
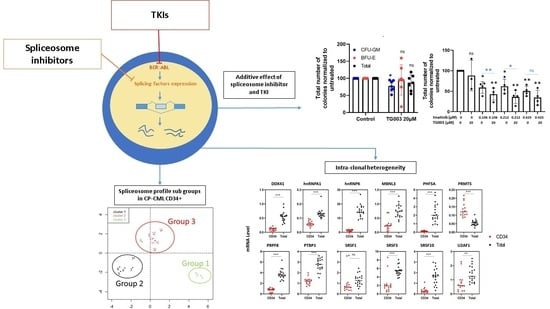
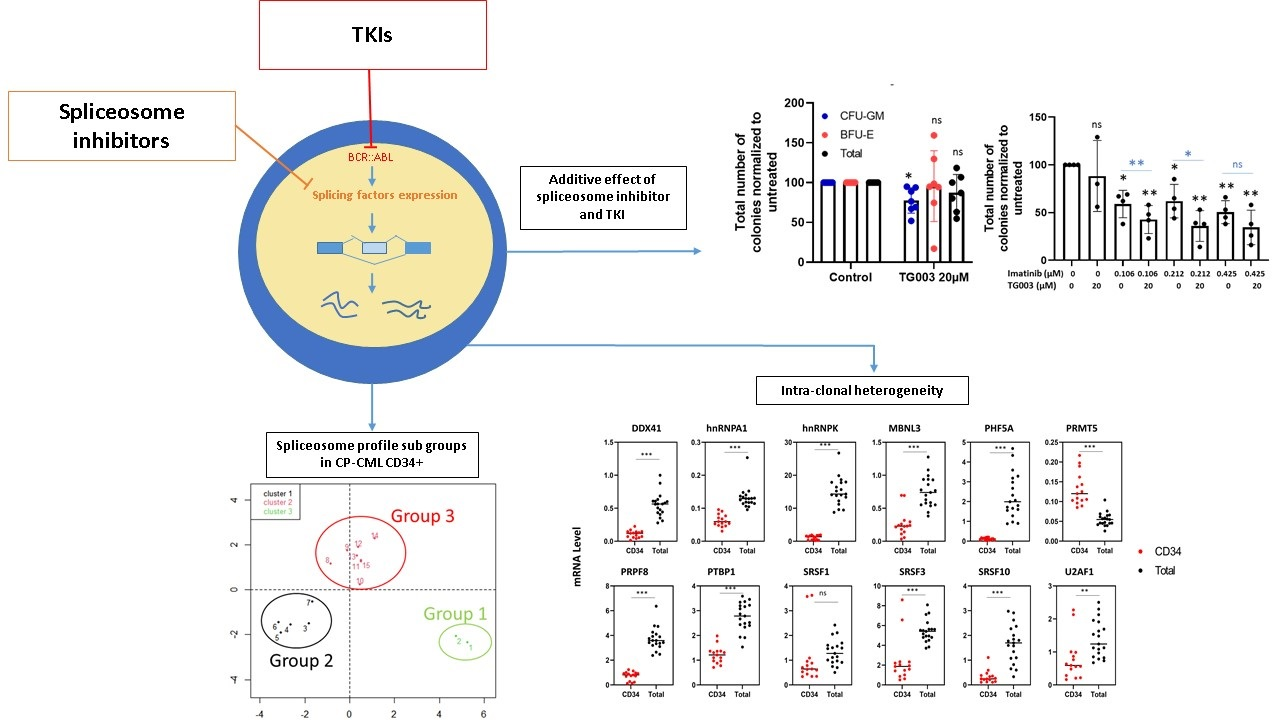
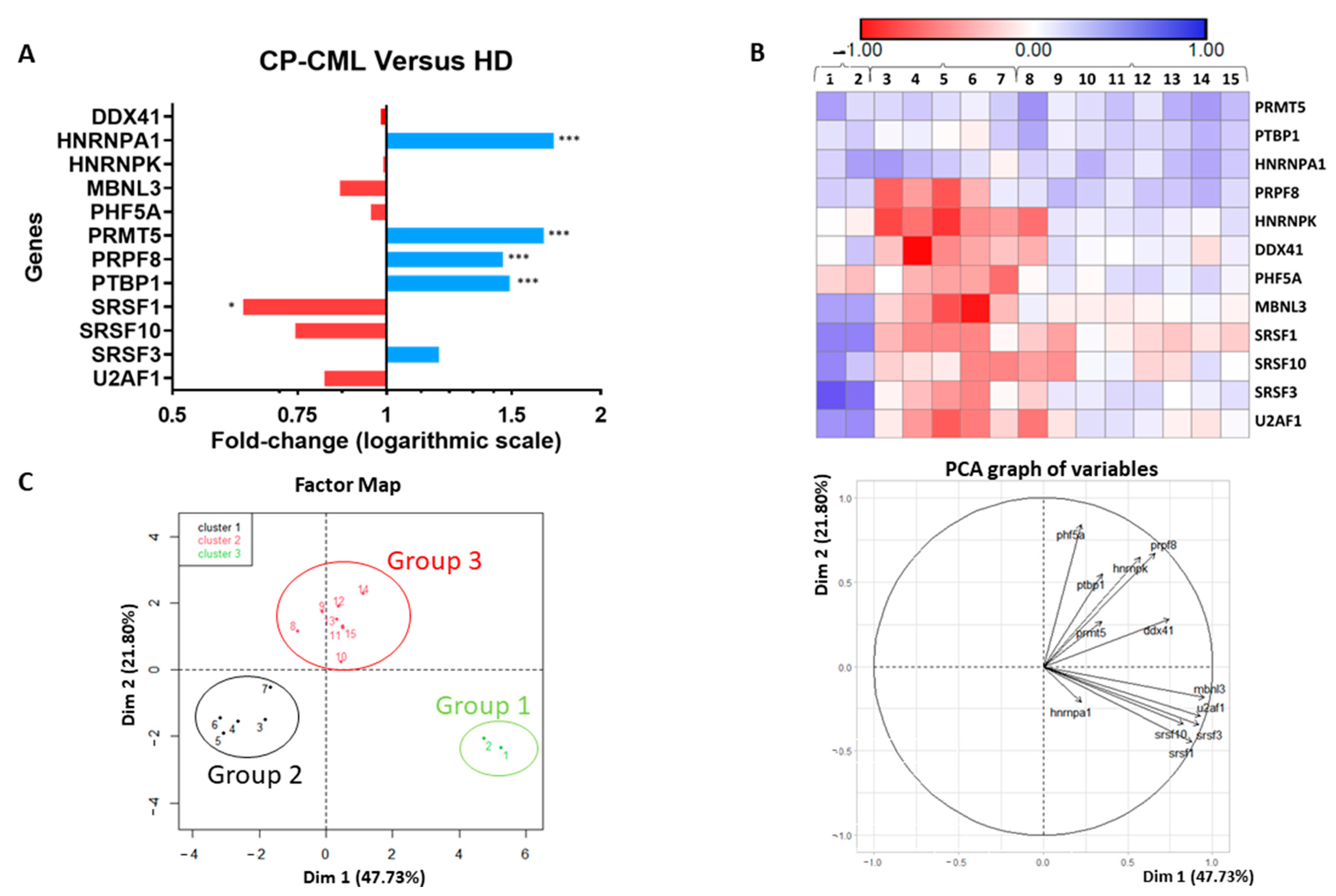
3.1. Splicing Genes Are Associated with Specific Signature Profiles
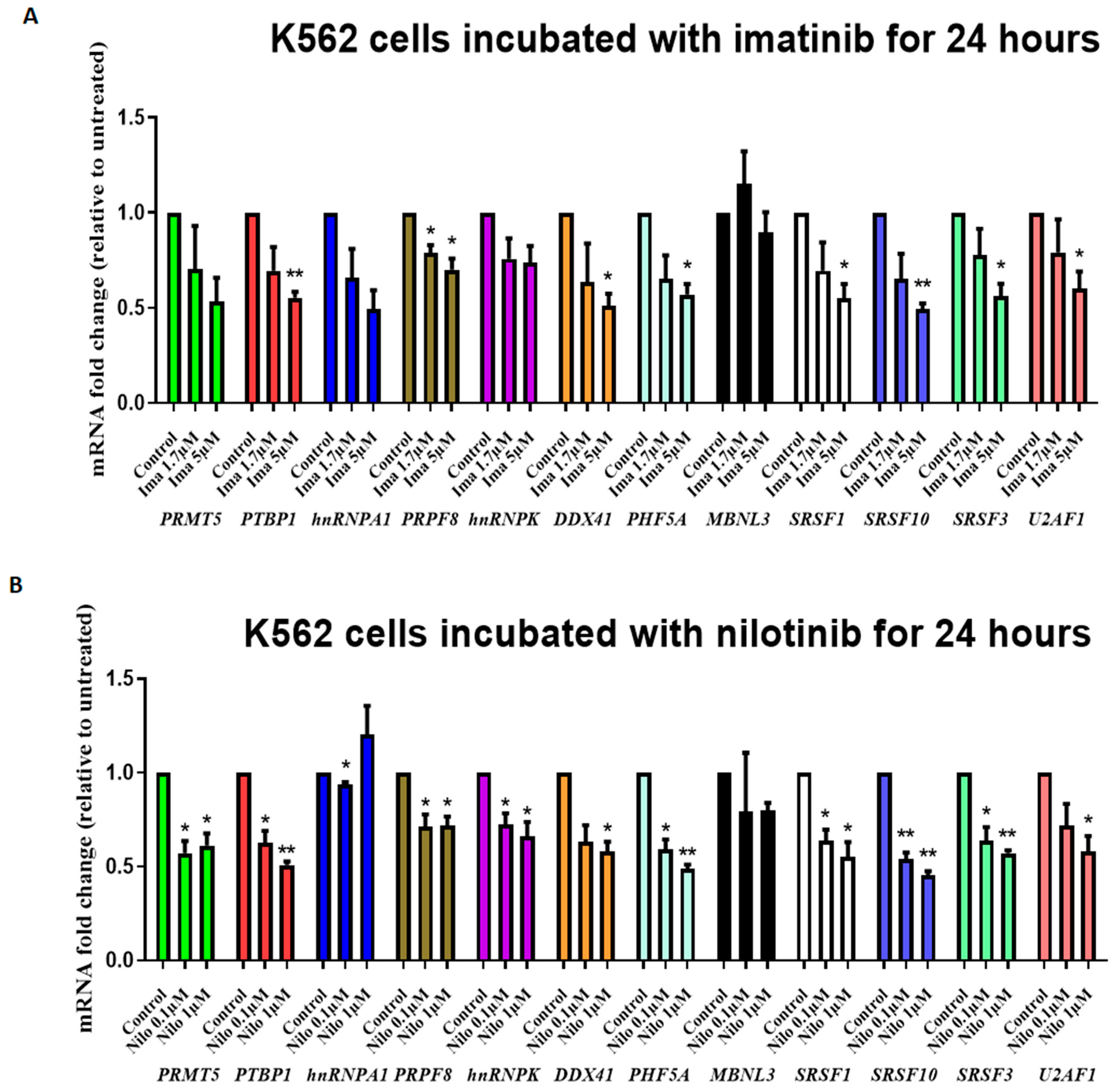
3.2. TKIs Affect Splicing Gene Expression
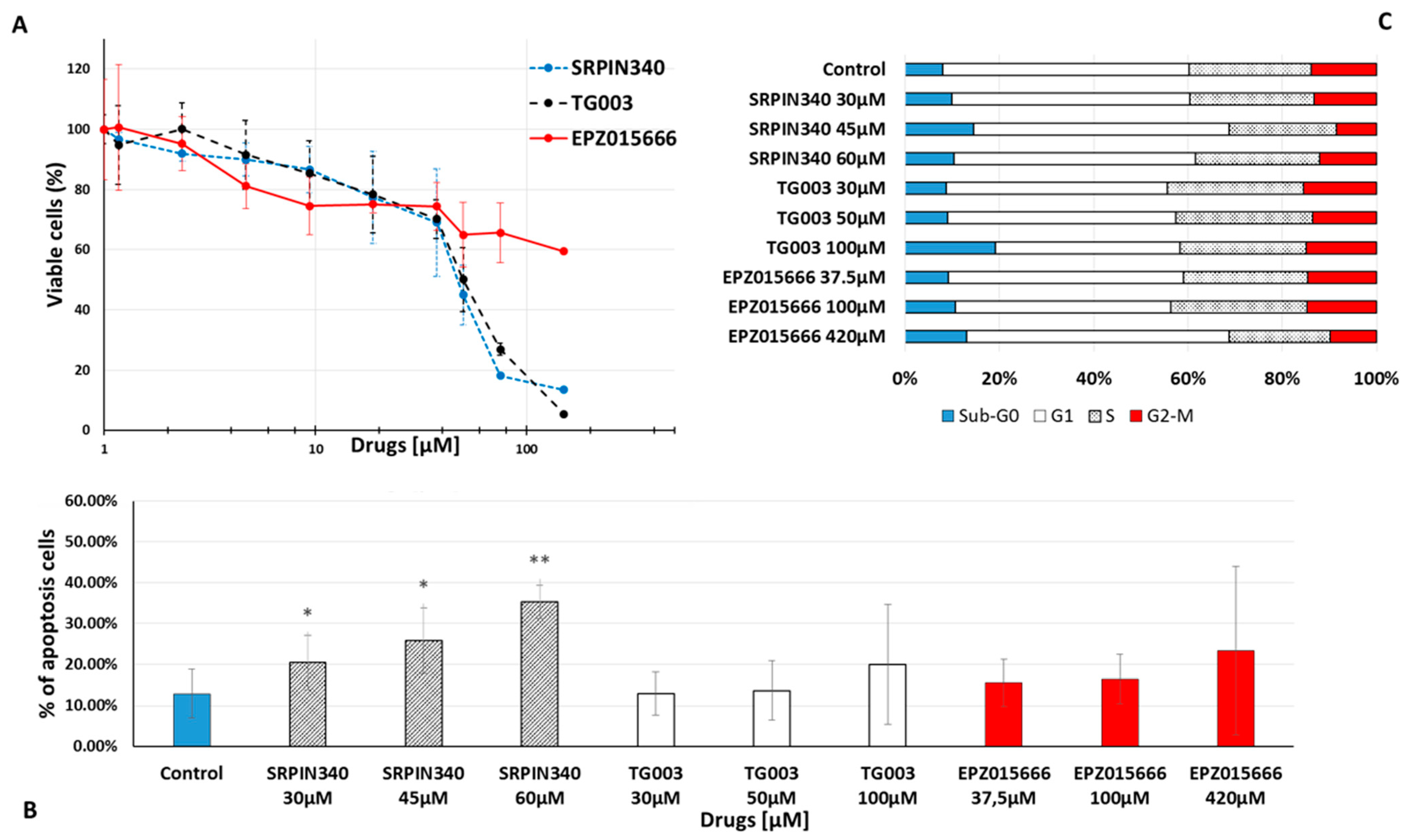
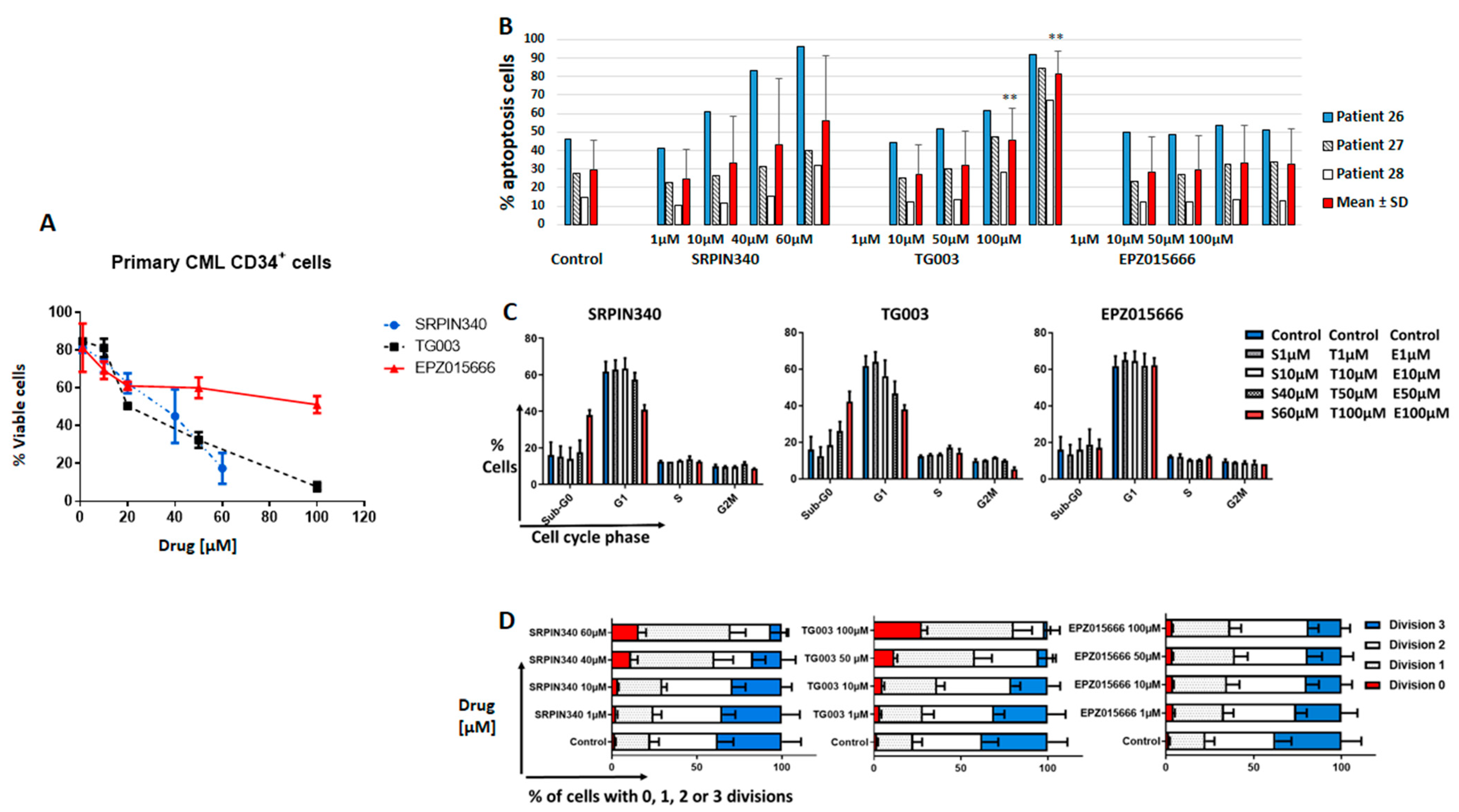
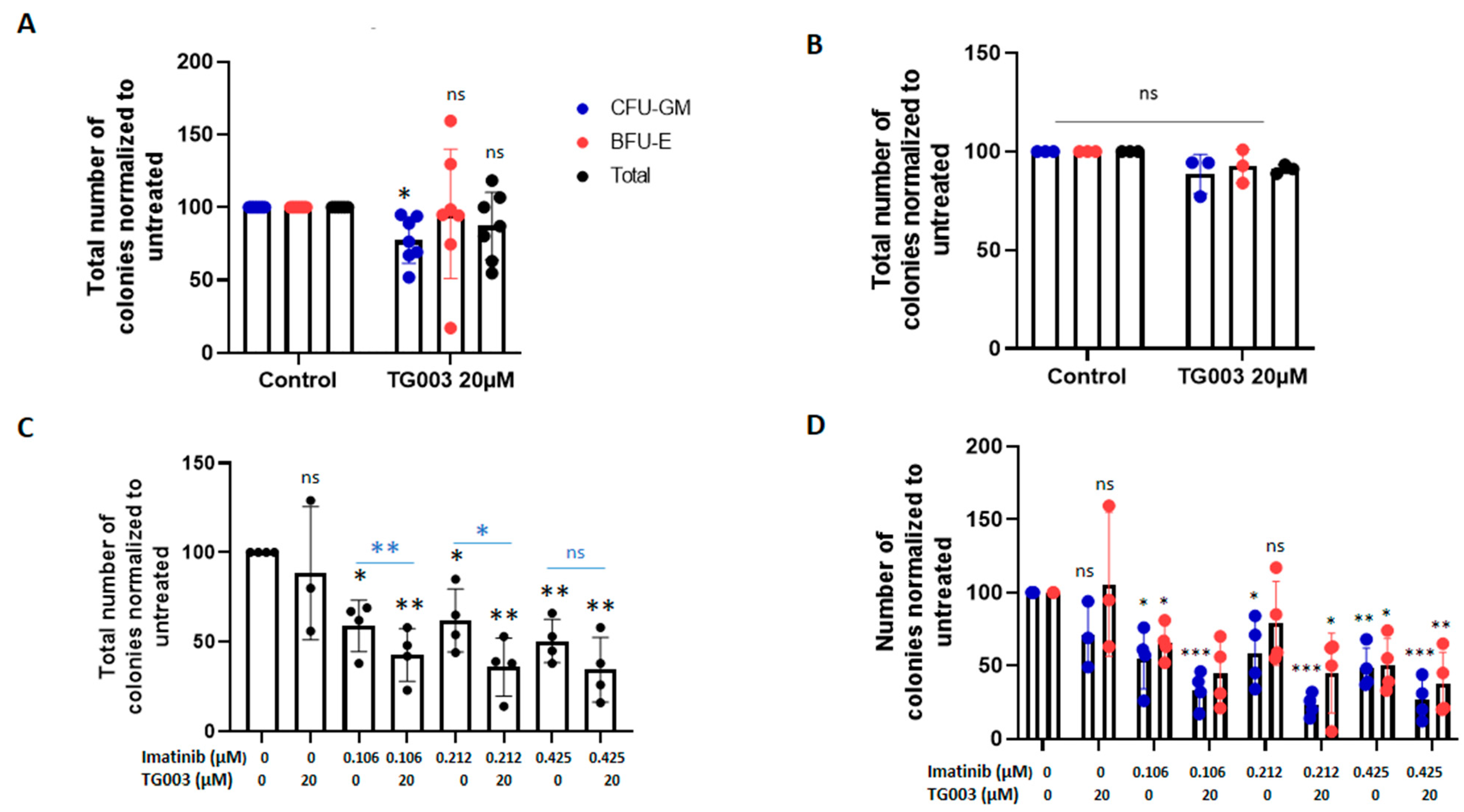
3.3. Effects of Spliceosome-Targeting Drugs in CML Cell Lines and Primary CP-CML CD34+ Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reimer, K.A.; Neugebauer, K.M. Blood Relatives: Splicing Mechanisms underlying Erythropoiesis in Health and Disease. F1000Research 2018, 7, 1364. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.; Vigevani, L.; Valcárcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef]
- Eymin, B. Targeting the spliceosome machinery: A new therapeutic axis in cancer? Biochem. Pharmacol. 2021, 189, 114039. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Maupetit-Mehouas, S.; Court, F.; Bourgne, C.; Guerci-Bresler, A.; Cony-Makhoul, P.; Johnson, H.; Etienne, G.; Rousselot, P.; Guyotat, D.; Janel, A.; et al. DNA methylation profiling reveals a pathological signature that contributes to transcriptional defects of CD34+CD15− cells in early chronic-phase chronic myeloid leukemia. Mol. Oncol. 2018, 12, 814–829. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Qian, J.; Yao, D.-M.; Qian, S.-X.; Qian, W.; Lin, J.; Xiao, G.-F.; Wang, C.-Z.; Deng, Z.-Q.; Ma, J.-C.; et al. SF3B1 mutation is a rare event in Chinese patients with acute and chronic myeloid leukemia. Clin. Biochem. 2013, 46, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Kubonishi, I.; Miyoshi, I. Establishment of a Ph1 chromosome-positive cell line from chronic myelogenous leukemia in blast crisis. Int. J. Cell Cloning 1983, 1, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 2016, 128, 1995. [Google Scholar] [CrossRef]
- Uzor, S.; Zorzou, P.; Bowler, E.; Porazinski, S.; Wilson, I.; Ladomery, M. Autoregulation of the human splice factor kinase CLK1 through exon skipping and intron retention. Gene 2018, 670, 46–54. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.-I.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Allende-Vega, N.; Dayal, S.; Agarwala, U.; Sparks, A.; Bourdon, J.C.; Saville, M.K. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 2013, 32, 1–14. [Google Scholar] [CrossRef]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef]
- Nishida, A.; Kataoka, N.; Takeshima, Y.; Yagi, M.; Awano, H.; Ota, M.; Itoh, K.; Hagiwara, M.; Matsuo, M. Chemical treatment enhances skipping of a mutated exon in the dystrophin gene. Nat. Commun. 2011, 2, 308. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.-P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Hagiwara, M. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef] [PubMed]
- Salesse, S.; Dylla, S.J.; Verfaillie, C.M. p210BCR/ABL-induced alteration of pre-mRNA splicing in primary human CD34+ hematopoietic progenitor cells. Leukemia 2004, 18, 727–733. [Google Scholar] [CrossRef]
- Karakama, Y.; Sakamoto, N.; Itsui, Y.; Nakagawa, M.; Tasaka-Fujita, M.; Nishimura-Sakurai, Y.; Kakinuma, S.; Oooka, M.; Azuma, S.; Tsuchiya, K.; et al. Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob. Agents Chemother. 2010, 54, 3179–3186. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.P.; Barbosa, É.d.A.A.; Polêto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.D.A.; De Oliveira, L.L.; Laranjeira, A.B.A.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.P.; Caetano, M.M.M.; de Souza, L.Â.; dos Passos, P.M.S.; Simaroli, N.B.; Barros, M.V.D.A.; de Souza, A.P.M.; de Oliveira, L.L.; Silva-Júnior, A.; Fietto, J.L.R.; et al. Combined SRPK and AKT pharmacological inhibition is synergistic in T-cell acute lymphoblastic leukemia cells. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2020, 65, 104777. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef]
- Yang, H.; Beutler, B.; Zhang, D. Emerging roles of spliceosome in cancer and immunity. Protein Cell 2022, 13, 559–579. Available online: https://link.springer.com/article/10.1007/s13238-021-00856-5 (accessed on 1 July 2022). [CrossRef]
- Brunner, A.M.; Steensma, D.P. Targeting Aberrant Splicing in Myelodysplastic Syndromes. Hematol. Oncol. Clin. 2020, 34, 379–391. [Google Scholar] [CrossRef]
- Li, S.-Q.; Liu, J.; Zhang, J.; Wang, X.-L.; Chen, D.; Wang, Y.; Xu, Y.-M.; Huang, B.; Lin, J.; Wang, X.-Z. Transcriptome profiling reveals the high incidence of hnRNPA1 exon 8 inclusion in chronic myeloid leukemia. J. Adv. Res. 2020, 24, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Sinnakannu, J.R.; Lee, K.L.; Cheng, S.; Li, J.; Yu, M.; Tan, S.P.; Ong, C.C.H.; Li, H.; Than, H.; Anczuków-Camarda, O.; et al. SRSF1 mediates cytokine-induced impaired imatinib sensitivity in chronic myeloid leukemia. Leukemia 2020, 34, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.A.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y.; et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wan, Z.; Wei, M.; Dong, Y.; Zhao, Y.; Chen, X.; Li, Z.; Qin, W.; Yang, G.; Liu, L. Chronic myelogenous leukemia cells remodel the bone marrow niche via exosome-mediated transfer of miR-320. Theranostics 2019, 9, 5642–5656. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Kumazaki, M.; Minami, Y.; Ito, Y.; Sugito, N.; Kuranaga, Y.; Taniguchi, K.; Yamada, N.; Otsuki, Y.; Naoe, T.; et al. Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 2016, 371, 1–11. [Google Scholar] [CrossRef]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, J.S.; Li, S.Q.; Badgett, T.C.; Song, Y.K.; Agarwal, S.; Coarfa, C.; Tolman, C.; Hurd, L.; Liao, H.; et al. MYCN controls an alternative RNA splicing program in high-risk metastatic neuroblastoma. Cancer Lett. 2016, 371, 214–224. [Google Scholar] [CrossRef]
- Hsu, T.Y.-T.; Simon, L.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.; Sun, T.; Kurley, S.; Tyagi, S.; et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Holm, F.; Hellqvist, E.; Mason, C.N.; Ali, S.A.; Delos-Santos, N.; Barrett, C.L.; Chun, H.-J.; Minden, M.D.; Moore, R.A.; Marra, M.A.; et al. Reversion to an embryonic alternative splicing program enhances leukemia stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2015, 112, 15444–15449. [Google Scholar] [CrossRef] [PubMed]
- Gullà, A.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Samur, M.K.; Qi, J.; Tai, Y.-T.; Harada, T.; Morelli, E.; Amodio, N.; et al. Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 2018, 32, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Ninomiya, K.; Okuno, Y.; Toyomoto, M.; Nishida, A.; Koike, Y.; Ohe, K.; Kii, I.; Yoshida, S.; Hashimoto, N.; et al. Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 46126. [Google Scholar] [CrossRef] [PubMed]
- Bowling, E.A.; Wang, J.H.; Gong, F.; Wu, W.; Neill, N.J.; Kim, I.S.; Tyagi, S.; Orellana, M.; Kurley, S.J.; Dominguez-Vidaña, R.; et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 2021, 184, 384–403.e21. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J.; Schattenberg, A.; Goldman, J.M.; Hertenstein, B.; Jacobsen, N.; Arcese, W.; Ljungman, P.; Ferrant, A.; Verdonck, L.; Niederwieser, D. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995, 86, 2041–2050. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102, 1368–1377. [Google Scholar] [CrossRef]
- Tarafdar, A.; Hopcroft, L.E.M.; Gallipoli, P.; Pellicano, F.; Cassels, J.; Hair, A.; Korfi, K.; Jørgensen, H.G.; Vetrie, D.; Holyoake, T.L.; et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 2017, 129, 199–208. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Change | FDR |
|---|---|---|
| PHF5A | 0.67 | 0.05 |
| PTBP1 | 0.74 | 0.02 |
| SRSF1 | 0.69 | 0.01 |
| SRSF3 | 0.67 | 0.01 |
| SRSF10 | 0.76 | 0.09 |
| Cell Line | SRPIN340 | TG003 | EPZ015666 |
|---|---|---|---|
| K562 (CML) | 45.2 ± 2.9 | 51.4 ± 14.4 | >100 |
| KCL22 (CML) | 57.5 ± 0.6 | 54.0 ± 13.5 | 96.1 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebecque, B.; Bourgne, C.; Munje, C.; Berger, J.; Tassin, T.; Cony-Makhoul, P.; Guerci-Bresler, A.; Johnson-Ansah, H.; Liu, W.; Saugues, S.; et al. The Spliceosome: A New Therapeutic Target in Chronic Myeloid Leukaemia. Cancers 2022, 14, 4695. https://doi.org/10.3390/cancers14194695
Lebecque B, Bourgne C, Munje C, Berger J, Tassin T, Cony-Makhoul P, Guerci-Bresler A, Johnson-Ansah H, Liu W, Saugues S, et al. The Spliceosome: A New Therapeutic Target in Chronic Myeloid Leukaemia. Cancers. 2022; 14(19):4695. https://doi.org/10.3390/cancers14194695
Chicago/Turabian StyleLebecque, Benjamin, Celine Bourgne, Chinmay Munje, Juliette Berger, Thomas Tassin, Pascale Cony-Makhoul, Agnès Guerci-Bresler, Hyacinthe Johnson-Ansah, Wei Liu, Sandrine Saugues, and et al. 2022. "The Spliceosome: A New Therapeutic Target in Chronic Myeloid Leukaemia" Cancers 14, no. 19: 4695. https://doi.org/10.3390/cancers14194695
APA StyleLebecque, B., Bourgne, C., Munje, C., Berger, J., Tassin, T., Cony-Makhoul, P., Guerci-Bresler, A., Johnson-Ansah, H., Liu, W., Saugues, S., Tchirkov, A., Vetrie, D., Copland, M., & Berger, M. G. (2022). The Spliceosome: A New Therapeutic Target in Chronic Myeloid Leukaemia. Cancers, 14(19), 4695. https://doi.org/10.3390/cancers14194695