
Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR


Abstract
:Highlights
- Two novel non-canonical splice site variants identified in MET genome.
- Predicted splicing strength using in silico splicing prediction tools.
- Tested routine cytological smear slides for RNA-based molecular diagnostics.
- RT-PCR and Sanger sequencing analysis confirmed MET exon 14 skipping.
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Sample Selection
2.2. Patient Samples, DNA and RNA Extraction
2.3. Cancer Hotspot Panel Library Preparation, Sequencing, and Data Analysis
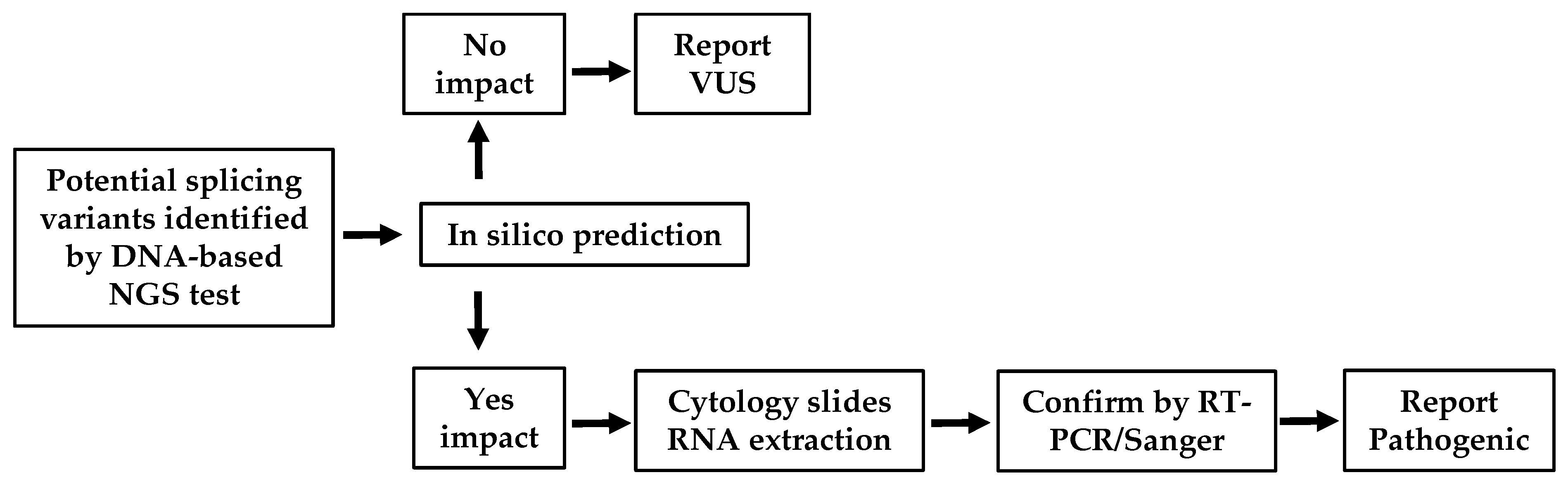
2.4. In Silico Prediction
2.5. RT-PCR and Sanger Sequencing
3. Results
3.1. Demographic and Clinical Characteristics of NSCLC Patients with Two Novel MET Variants
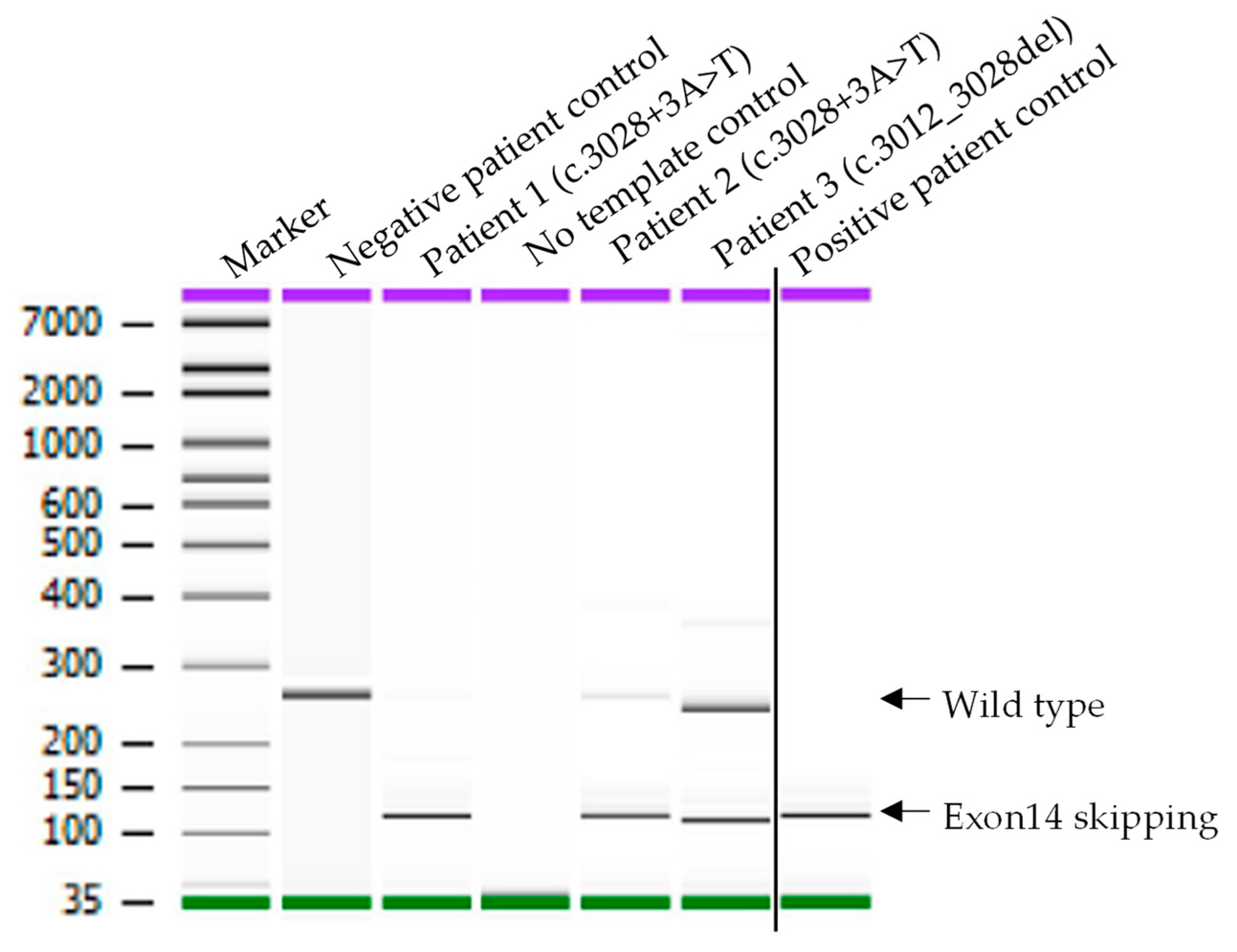
3.2. Confirmation of MET Exon 14 Mutation in Two Novel MET Variants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Zaric, B.; Rabea, A.; Thongprasert, S.; Lertprasertsuke, N.; Dalurzo, M.L.; Varella-Garcia, M. Biomarker Testing for Personalized Therapy in Lung Cancer in Low- and Middle-Income Countries. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoda, S.; Dagogo-Jack, I.; Hata, A.N. Targeting oncogenic drivers in lung cancer: Recent progress, current challenges and future opportunities. Pharmacol. Ther. 2019, 193, 20–30. [Google Scholar] [CrossRef]
- Vuong, H.G.; Ho, A.T.N.; Altibi, A.M.A.; Nakazawa, T.; Katoh, R.; Kondo, T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer—A systematic review and meta-analysis. Lung Cancer 2018, 123, 76–82. [Google Scholar] [CrossRef]
- NIH. National Cancer Institute: Drugs Approved for Lung Cancer. 2021. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/lung (accessed on 7 February 2022).
- TABRECTA (Capmatinib) Tablets: How TABRECTA May Help. 2021. Available online: https://www.us.tabrecta.com/met-exon-14-skipping-mutation-nsclc/about-tabrecta/how-tabrecta-may-help/ (accessed on 7 February 2022).
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Thorac. Oncol. 2018, 13, 323–358. [Google Scholar] [PubMed] [Green Version]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.H.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Suda, K.; Yatabe, Y. Surgery for NSCLC in the era of personalized medicine. Nat. Rev. Clin. Oncol. 2013, 10, 235–244. [Google Scholar] [CrossRef]
- Xu, Z.; Li, H.; Dong, Y.; Cheng, P.; Luo, F.; Fu, S.; Gao, M.; Kong, L.; Che, N. Incidence and PD-L1 Expression of MET 14 Skipping in Chinese Population: A Non-Selective NSCLC Cohort Study Using RNA-Based Sequencing. OncoTargets Ther. 2020, 13, 6245–6253. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Peled, N.; Greer, J.; Wu, W.; Choi, P.; Berger, A.H.; Wong, S.; Jen, K.-Y.; Seo, Y.; Hann, B.; et al. MET Exon 14 Mutation Encodes an Actionable Therapeutic Target in Lung Adenocarcinoma. Cancer Res. 2017, 77, 4498–4505. [Google Scholar] [CrossRef] [Green Version]
- Awad, M.M. Impaired c-Met Receptor Degradation Mediated by MET Exon 14 Mutations in Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 879–881. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, 653–663. [Google Scholar] [CrossRef]
- Shi, M.; Ma, J.; Feng, M.; Liang, L.; Chen, H.; Wang, T.; Xie, Z. Novel MET exon 14 skipping analogs characterized in non-small cell lung cancer patients: A case study. Cancer Genet. 2021, 256–257, 62–67. [Google Scholar] [CrossRef]
- Reynolds, J.P.; Zhou, Y.; Jakubowski, M.A.; Wang, Z.; Brainard, J.A.; Klein, R.D.; Farver, C.F.; Almeida, F.A.; Cheng, Y.-W. Next-generation sequencing of liquid-based cytology non-small cell lung cancer samples. Cancer Cytopathol. 2017, 125, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.W.; Stefaniuk, C.; Jakubowski, M.A. Real-time PCR and targeted next-generation sequencing in the detection of low level EGFR mutations: Instructive case analyses. Respir. Med. Case Rep. 2019, 28, 100901. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Geiersbach, K.; Chadwick, B. Rapid removal of cytology slide coverslips for DNA and RNA isolation. J. Am. Soc. Cytopathol. 2017, 6, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Oxnard, G.R.; Elkin, S.; Sullivan, E.K.; Carter, J.L.; Barbie, D.A. Response to Crizotinib in a Patient With Lung Adenocarcinoma Harboring a MET Splice Site Mutation. Clin. Lung Cancer 2015, 16, e101–e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendenhall, M.A.; Goldman, J.W. MET-Mutated NSCLC with Major Response to Crizotinib. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, e33–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafrir, Z.; Tuller, T. Nucleotide sequence composition adjacent to intronic splice sites improves splicing efficiency via its effect on pre-mRNA local folding in fungi. RNA 2015, 21, 1704–1718. [Google Scholar] [CrossRef] [Green Version]
- Fairbrother, W.G.; Yeo, G.W.; Yeh, R.; Goldstein, P.; Mawson, M.; Sharp, P.A.; Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004, 32, W187–W190. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.X.; Zhang, M.; Krainer, A.R. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998, 12, 1998–2012. [Google Scholar] [CrossRef] [Green Version]
- Schaal, T.D.; Maniatis, T. Selection and characterization of pre-mRNA splicing enhancers: Identification of novel SR protein-specific enhancer sequences. Mol. Cell. Biol. 1999, 19, 1705–1719. [Google Scholar] [CrossRef] [Green Version]
- Coulter, L.R.; Landree, M.A.; Cooper, T.A. Identification of a new class of exonic splicing enhancers by in vivo selection. Mol. Cell. Biol. 1997, 17, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Kangsamaksin, T.; Chao, M.S.; Banerjee, J.K.; Chasin, L.A. Exon inclusion is dependent on predictable exonic splicing enhancers. Mol. Cell. Biol. 2005, 25, 7323–7332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.H.; Leslie, C.S.; Chasin, L.A. Computational searches for splicing signals. Methods 2005, 37, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jia, Y.; Stoopler, M.B.; Shen, Y.; Cheng, H.; Chen, J.; Mansukhani, M.; Koul, S.; Halmos, B.; Borczuk, A.C. Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkiewicz, M.; Saqi, A.; Mansukhani, M.M.; Hodel, V.; Krull, A.; Shu, C.A.; Fernandes, H.D. Efficacy of DNA versus RNA NGS-based Methods in MET Exon 14 skipping mutation detection. J. Clin. Oncol. 2020, 38, 9036. [Google Scholar] [CrossRef]
- Gentien, D.; Piqueret-Stephan, L.; Henry, E.; Albaud, B.; Rapinat, A.; Koscielny, S.; Scoazec, J.-Y.; Vielh, P. Digital Multiplexed Gene Expression Analysis of mRNA and miRNA from Routinely Processed and Stained Cytological Smears: A Proof-of-Principle Study. Acta Cytol. 2021, 65, 88–98. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Histology | Adenocarcinoma | Adenocarcinoma | Necrotic NSCLC |
| Tumor% | 40 | 80 | 90 |
| MET variant NG_008996.1 (NM_000245.3) | c.3028+3A>T VAF = 24% NGS read depth = 3761 | c.3028+3A>T VAF = 37% NGS read depth = 10,184 | c.3012_3028del VAF = 11% NGS read depth = 3518 |
| Other activating mutations in hotspots of BRAF, EGFR, HER2, and KRAS | Negative | KRAS(NM_004985.3) c.34G>T (p.Gly12Cys) | Negative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, R.; Jakubowski, M.A.; Spildener, J.; Cheng, Y.-W. Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR. Cancers 2022, 14, 4814. https://doi.org/10.3390/cancers14194814
Das R, Jakubowski MA, Spildener J, Cheng Y-W. Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR. Cancers. 2022; 14(19):4814. https://doi.org/10.3390/cancers14194814
Chicago/Turabian StyleDas, Riku, Maureen A. Jakubowski, Jessica Spildener, and Yu-Wei Cheng. 2022. "Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR" Cancers 14, no. 19: 4814. https://doi.org/10.3390/cancers14194814
APA StyleDas, R., Jakubowski, M. A., Spildener, J., & Cheng, Y. -W. (2022). Identification of Novel MET Exon 14 Skipping Variants in Non-Small Cell Lung Cancer Patients: A Prototype Workflow Involving in Silico Prediction and RT-PCR. Cancers, 14(19), 4814. https://doi.org/10.3390/cancers14194814