

Gene Regulatory Network Characterization of Gastric Cancer’s Histological Subtypes: Distinctive Biological and Clinically Relevant Master Regulators

 ,
,  , , ,
, , ,  , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Patient Selection and Study Design
2.3. Data Processing
- Signet-Ring Cells (SRC),
- Diffuse (poorly cohesive, not SRC)
- Intestinal mucinous,
- Intestinal papillary,
- Intestinal tubular,
- Intestinal not otherwise specified (iNOS),
- Stomach not otherwise specified (sNOS).
- Diffuse (poorly cohesive, not SRC),
- Intestinal (tubular and iNOS).
2.4. Statistical Analysis
2.4.1. Association with Clinical Features and Survival Analyses
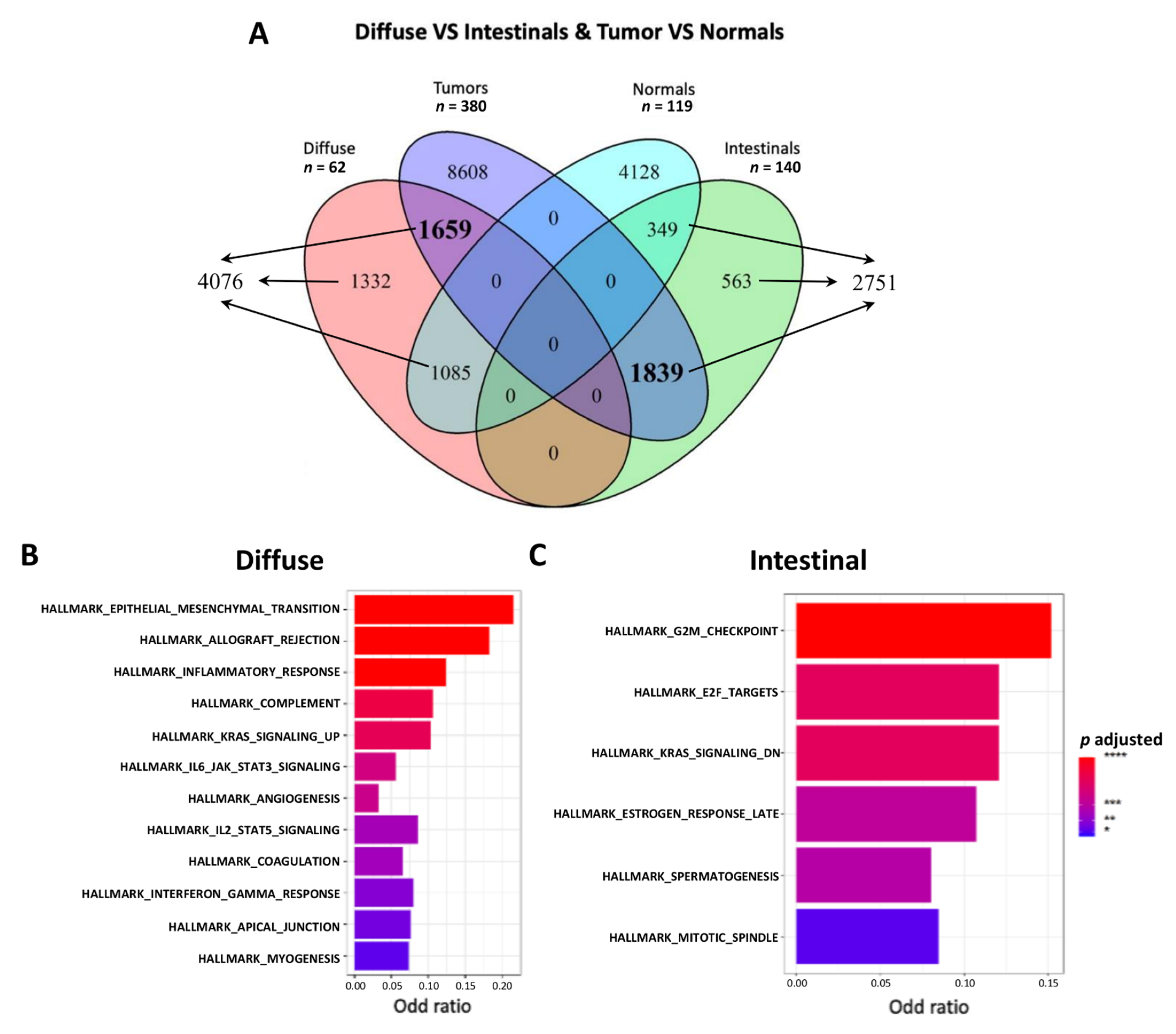
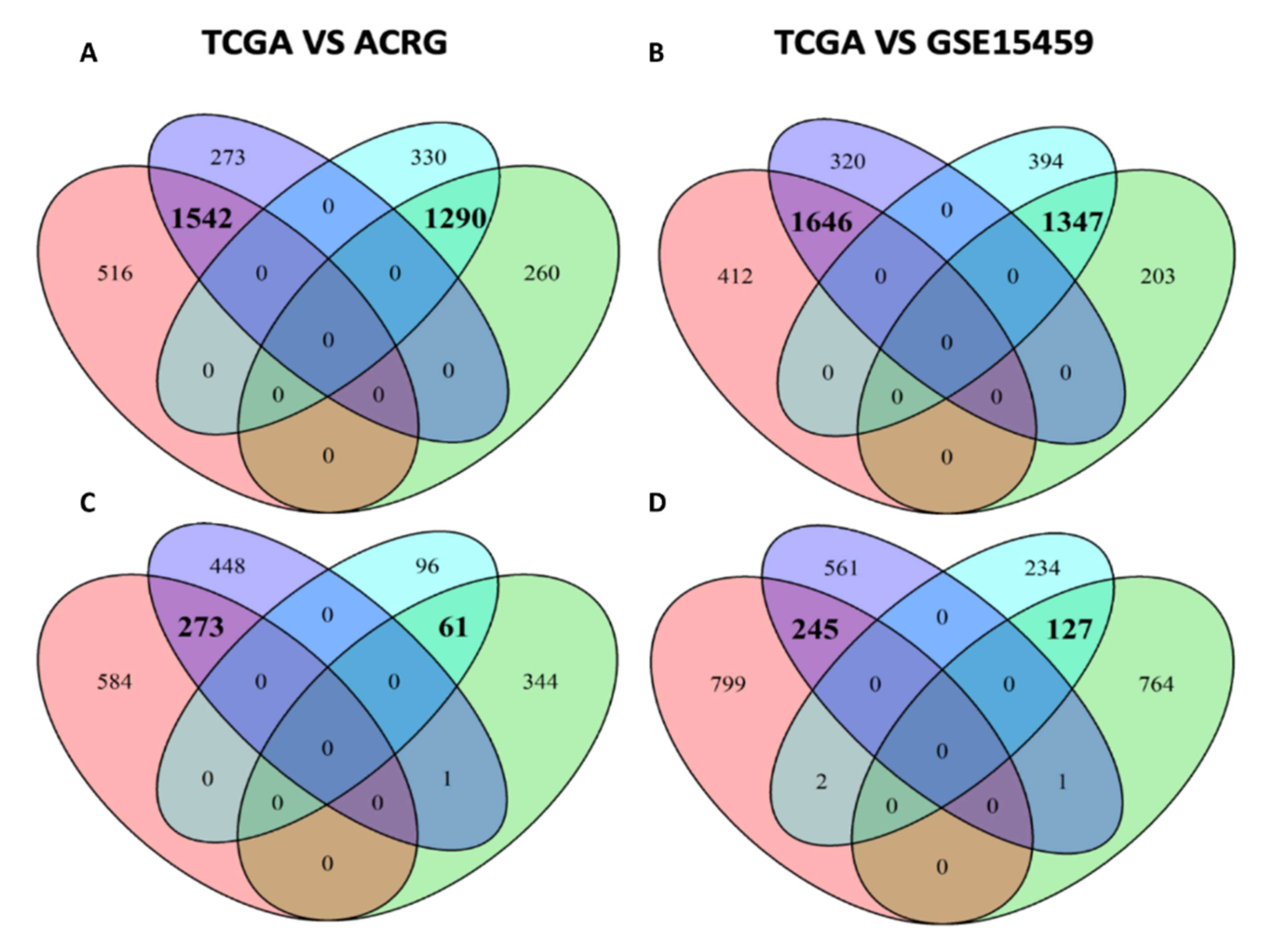
2.4.2. Differential Expression Analysis and Venn
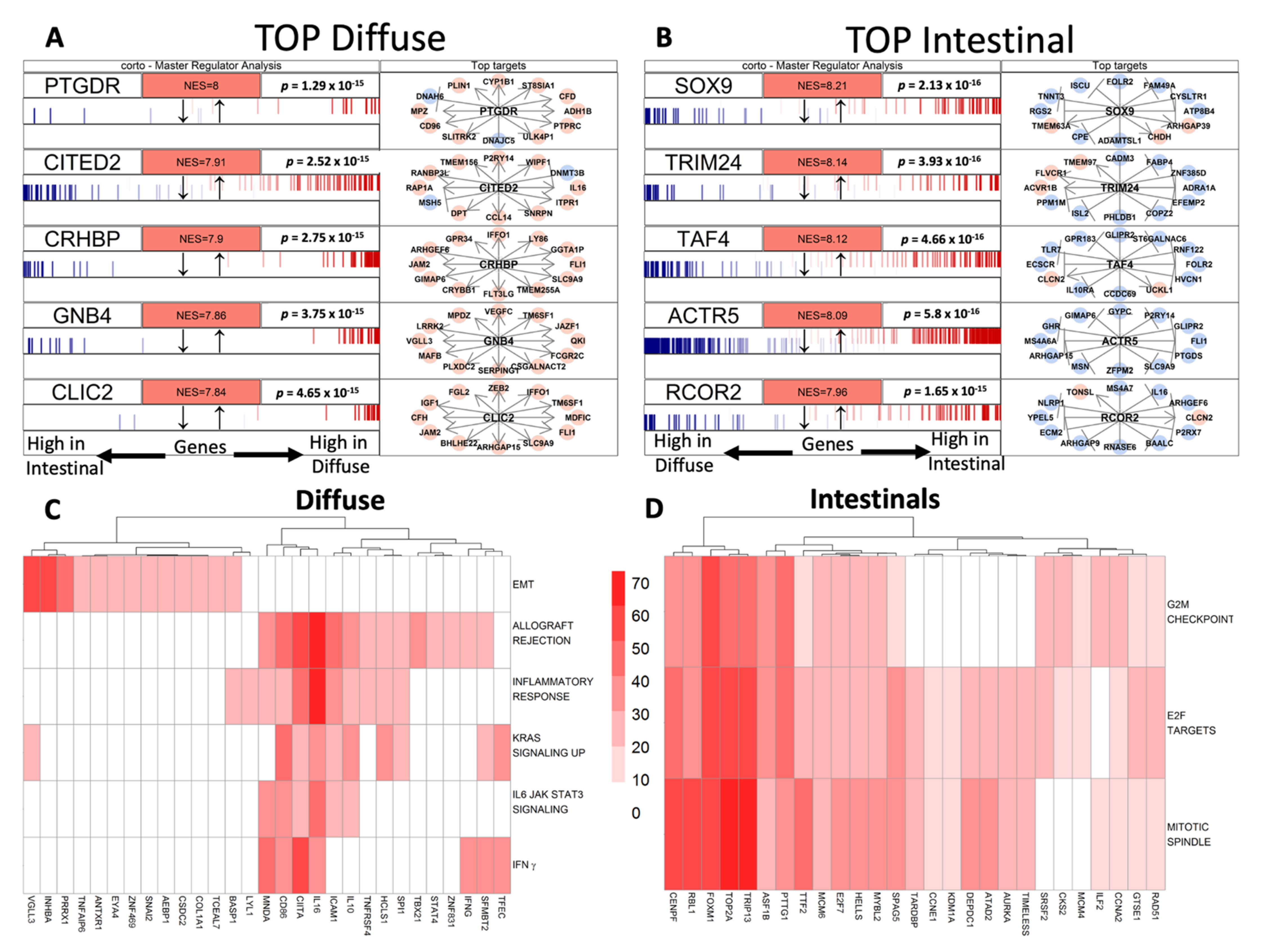
2.4.3. Gene-Set Enrichment Analyses and Master Regulator Analyses (MRA)
2.4.4. Single Sample Gene Set Enrichment Analysis (ssGSEA) and Single Sample Master Regulator Analysis (ssMRA)
2.4.5. Association of MRs with Clinical and Survival Features
3. Results
3.1. In Silico-Refined Histological Subtypes Have Similar Clinicopathological Characteristics
3.2. Functional Enrichment Highlighted a Different Biological Behavior for the Two Histological Subtypes
3.3. Gene Regulatory Networks Highlights Different Putative Hub Genes
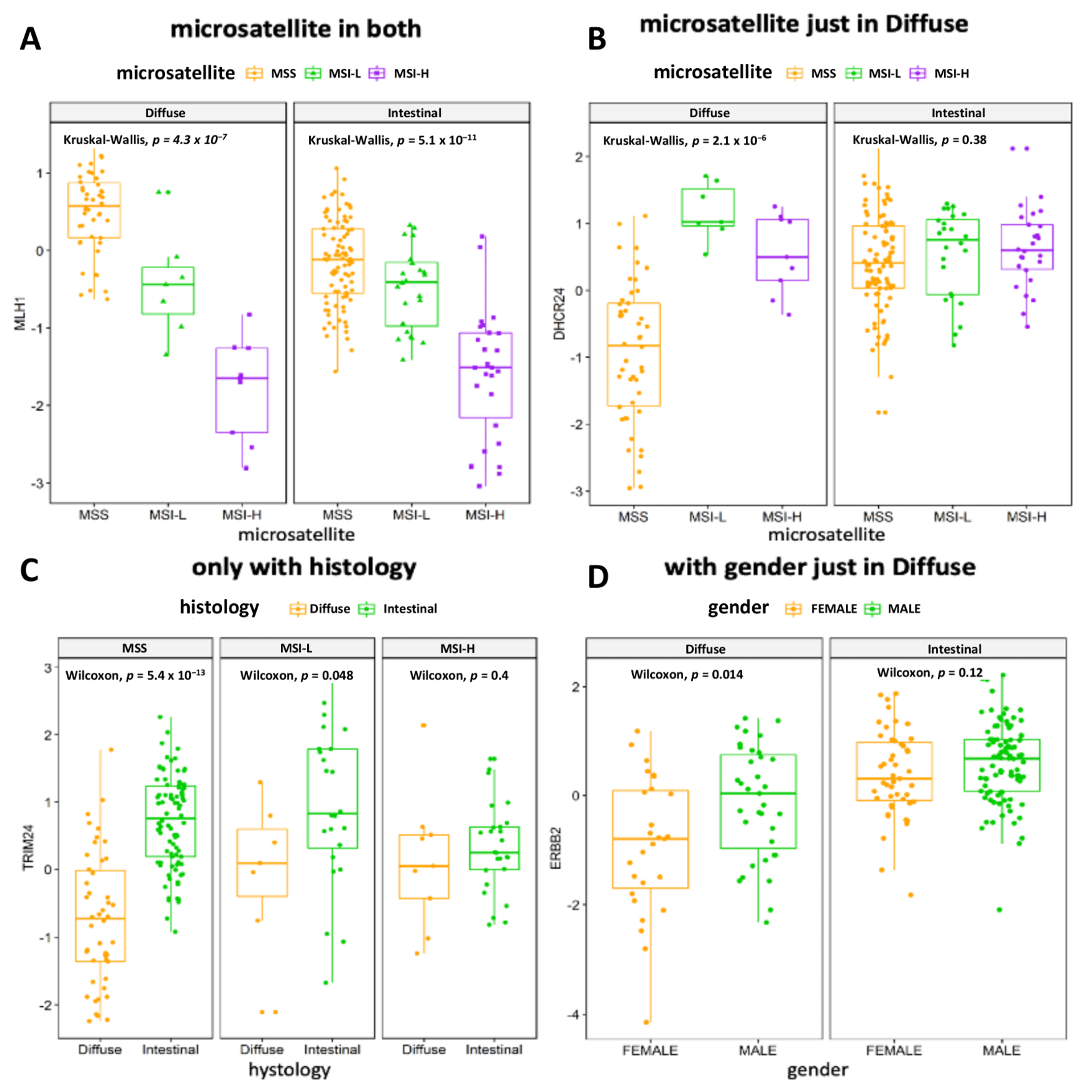
3.4. The Association of MRs Activity with Clinical Variables or Prognosis Confirms the Relevance of Underlying Molecular Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.R.; Allum, W.H. Gastric tumours. Medicine 2019, 47, 309–313. [Google Scholar] [CrossRef]
- Cisło, M.; Filip, A.A.; Arnold Offerhaus, G.J.; Ciseł, B.; Rawicz-Pruszyński, K.; Skierucha, M.; Polkowski, W.P. Distinct molecular subtypes of gastric cancer: From Laurén to molecular pathology. Oncotarget 2018, 9, 19427–19442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Fléjou, J.-F. [WHO Classification of digestive tumors: The fourth edition]. Ann. Pathol. 2011, 31, S27–S31. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Böger, C.; Krüger, S.; Behrens, H.M.; Bock, S.; Haag, J.; Kalthoff, H.; Röcken, C. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann. Oncol. 2017, 28, 1005–1014. [Google Scholar] [CrossRef]
- Lee, H.H.; Kim, S.Y.; Jung, E.S.; Yoo, J.; Kim, T.-M. Mutation heterogeneity between primary gastric cancers and their matched lymph node metastases. Gastric Cancer 2019, 22, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Mathiak, M.; Warneke, V.S.; Behrens, H.-M.; Haag, J.; Böger, C.; Krüger, S.; Röcken, C. Clinicopathologic characteristics of microsatellite instable gastric carcinomas revisited: Urgent need for standardization. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 12–24. [Google Scholar] [CrossRef]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polom, K.; Böger, C.; Smyth, E.; Marrelli, D.; Behrens, H.-M.; Marano, L.; Becker, T.; Lordick, F.; Röcken, C.; Roviello, F. Synchronous metastatic gastric cancer-molecular background and clinical implications with special attention to mismatch repair deficiency. Eur. J. Surg. Oncol. 2018, 44, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, D.; Polom, K.; Pascale, V.; Vindigni, C.; Piagnerelli, R.; De Franco, L.; Ferrara, F.; Roviello, G.; Garosi, L.; Petrioli, R.; et al. Strong Prognostic Value of Microsatellite Instability in Intestinal Type Non-cardia Gastric Cancer. Ann. Surg. Oncol. 2016, 23, 943–950. [Google Scholar] [CrossRef]
- Marrelli, D.; Polom, K.; Neri, A.; Roviello, F. Clinical impact of molecular classifications in gastric cancer. Updates Surg. 2018, 70, 225–232. [Google Scholar] [CrossRef]
- Polom, K.; Marano, L.; Marrelli, D.; De Luca, R.; Roviello, G.; Savelli, V.; Tan, P.; Roviello, F. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br. J. Surg. 2018, 105, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sohn, B.H.; Hwang, J.-E.; Jang, H.-J.; Lee, H.-S.; Oh, S.C.; Shim, J.-J.; Lee, K.-W.; Kim, E.H.; Yim, S.Y.; Lee, S.H.; et al. Clinical significance of four molecular subtypes of gastric cancer identified by the cancer genome atlas project. Clin. Cancer Res. 2017, 23, 4441–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Xie, Q.; Liu, Y.; Guo, H.; Ren, Y.; Li, J.; Zhao, Q. Clinical characteristics and prognostic significance of TCGA and ACRG classification in gastric cancer among the Chinese population. Mol. Med. Rep. 2020, 22, 828–840. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Min, L.; Zhao, Y.; Zhu, S.; Qiu, X.; Cheng, R.; Xing, J.; Shao, L.; Guo, S.; Zhang, S. Integrated analysis identifies molecular signatures and specific prognostic factors for different gastric cancer subtypes. Transl. Oncol. 2017, 10, 99–107. [Google Scholar] [CrossRef]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippe, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, C.H.; Ivanova, T.; Wu, J.; Lee, M.; Tan, I.B.; Tao, J.; Ward, L.; Koo, J.H.; Gopalakrishnan, V.; Zhu, Y.; et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009, 5, e1000676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russi, S.; Calice, G.; Ruggieri, V.; Laurino, S.; La Rocca, F.; Amendola, E.; Lapadula, C.; Compare, D.; Nardone, G.; Musto, P.; et al. Gastric normal adjacent mucosa versus healthy and cancer tissues: Distinctive transcriptomic profiles and biological features. Cancers 2019, 11, 1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariette, C.; Carneiro, F.; Grabsch, H.I.; van der Post, R.S.; Allum, W.; de Manzoni, G. European Chapter of International Gastric Cancer Association Consensus on the pathological definition and classification of poorly cohesive gastric carcinoma. Gastric Cancer 2019, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; El Hajj, N.; Sittler, S.; Lammert, N.; Barnes, R.; Meloni-Ehrig, A. Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 2012, 3, 251–261. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mercatelli, D.; Ray, F.; Giorgi, F.M. Pan-Cancer and Single-Cell Modeling of Genomic Alterations Through Gene Expression. Front Genet. 2019, 10, 671. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, F.M.; Lachmann, A.; Lopez, G.; Califano, A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 2016, 32, 2233–2235. [Google Scholar] [CrossRef]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. corto: A lightweight R package for gene network inference and master regulator analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016; p. 276. ISBN 978-3-319-24277-4. [Google Scholar]
- Xie, D.; Pei, Q.; Li, J.; Wan, X.; Ye, T. Emerging role of E2F family in cancer stem cells. Front. Oncol. 2021, 11, 723137. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, R.-L.; Xu, A.-M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 2015, 7, 2141–2158. [Google Scholar]
- Chandanos, E.; Rubio, C.A.; Lindblad, M.; Jia, C.; Tsolakis, A.V.; Warner, M.; Gustafsson, J.-A.; Lagergren, J. Endogenous estrogen exposure in relation to distribution of histological type and estrogen receptors in gastric adenocarcinoma. Gastric Cancer 2008, 11, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; Alvarez, M.J.; Talos, F.; Dhruv, H.; Rieckhof, G.E.; Iyer, A.; Diefes, K.L.; Aldape, K.; Berens, M.; Shen, M.M.; et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 2014, 159, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, L.; Mao, Y.; Hua, D. VGLL3 is a prognostic biomarker and correlated with clinical pathologic features and immune infiltrates in stomach adenocarcinoma. Sci. Rep. 2020, 10, 1355. [Google Scholar] [CrossRef] [Green Version]
- Townson, S.M.; Dobrzycka, K.M.; Lee, A.V.; Air, M.; Deng, W.; Kang, K.; Jiang, S.; Kioka, N.; Michaelis, K.; Oesterreich, S. SAFB2, a new scaffold attachment factor homolog and estrogen receptor corepressor. J. Biol. Chem. 2003, 278, 20059–20068. [Google Scholar] [CrossRef] [Green Version]
- Roviello, F.; Marano, L.; Ambrosio, M.R.; Resca, L.; D’Ignazio, A.; Petrelli, F.; Petrioli, R.; Costantini, M.; Polom, K.; Macchiarelli, R.; et al. Signet ring cell percentage in poorly cohesive gastric cancer patients: A potential novel predictor of survival. Eur. J. Surg. Oncol. 2022, 48, 561–569. [Google Scholar] [CrossRef]
- Marrelli, D.; Pedrazzani, C.; Morgagni, P.; de Manzoni, G.; Pacelli, F.; Coniglio, A.; Marchet, A.; Saragoni, L.; Giacopuzzi, S.; Roviello, F.; et al. Changing clinical and pathological features of gastric cancer over time. Br. J. Surg. 2011, 98, 1273–1283. [Google Scholar] [CrossRef]
- Henson, D.E.; Dittus, C.; Younes, M.; Nguyen, H.; Albores-Saavedra, J. Differential trends in the intestinal and diffuse types of gastric carcinoma in the United States, 1973-2000: Increase in the signet ring cell type. Arch. Pathol. Lab. Med. 2004, 128, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Amorosi, A.; Palli, D. Epidemiology of intestinal and difuse types of gastric carcinoma: A time-trend study in Finland with comparison between studies from high- and low-risk areas. Cancer 1994, 73, 1533. [Google Scholar] [CrossRef]
- Marrelli, D.; Polom, K.; de Manzoni, G.; Morgagni, P.; Baiocchi, G.L.; Roviello, F. Multimodal treatment of gastric cancer in the west: Where are we going? World J. Gastroenterol. 2015, 21, 7954–7969. [Google Scholar] [CrossRef] [PubMed]
- Hori, N.; Takakura, Y.; Sugino, A.; Iwasawa, S.; Nomizo, K.; Yamaguchi, N.; Takano, H.; Yamaguchi, N. Vestigial-like family member 3 stimulates cell motility by inducing high-mobility group AT-hook 2 expression in cancer cells. J. Cell. Mol. Med. 2022, 26, 2686–2697. [Google Scholar] [CrossRef]
- Zhou, W.; Gross, K.M.; Kuperwasser, C. Molecular regulation of Snai2 in development and disease. J. Cell Sci. 2019, 132, jcs235127. [Google Scholar] [CrossRef]
- Hill, D.G.; Yu, L.; Gao, H.; Balic, J.J.; West, A.; Oshima, H.; McLeod, L.; Oshima, M.; Gallimore, A.; D’Costa, K.; et al. Hyperactive gp130/STAT3-driven gastric tumourigenesis promotes submucosal tertiary lymphoid structure development. Int. J. Cancer 2018, 143, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Hippo, Y.; Yashiro, M.; Ishii, M.; Taniguchi, H.; Tsutsumi, S.; Hirakawa, K.; Kodama, T.; Aburatani, H. Differential gene expression profiles of scirrhous gastric cancer cells with high metastatic potential to peritoneum or lymph nodes. Cancer Res. 2001, 61, 889–895. [Google Scholar]
- Satoh, A.; Toyota, M.; Ikeda, H.; Morimoto, Y.; Akino, K.; Mita, H.; Suzuki, H.; Sasaki, Y.; Kanaseki, T.; Takamura, Y.; et al. Epigenetic inactivation of class II transactivator (CIITA) is associated with the absence of interferon-gamma-induced HLA-DR expression in colorectal and gastric cancer cells. Oncogene 2004, 23, 8876–8886. [Google Scholar] [CrossRef] [Green Version]
- Osaki, L.H.; Bockerstett, K.A.; Wong, C.F.; Ford, E.L.; Madison, B.B.; DiPaolo, R.J.; Mills, J.C. Interferon-γ directly induces gastric epithelial cell death and is required for progression to metaplasia. J. Pathol. 2019, 247, 513–523. [Google Scholar] [CrossRef]
- Richmond, J.; Tuzova, M.; Cruikshank, W.; Center, D. Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J. Cell. Physiol. 2014, 229, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Chu, H.-J.; Li, N.; Huang, L.-Y.; Chen, J. Centromere Protein I (CENP-I) Is Upregulated in Gastric Cancer, Predicts Poor Prognosis, and Promotes Tumor Cell Proliferation and Migration. Technol. Cancer Res. Treat. 2021, 20, 15330338211045510. [Google Scholar] [CrossRef]
- Millour, J.; de Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W.F. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Li, X.; Zhang, Y.; Yao, R.; Qiu, W.; Tang, D.; Liang, J. Overexpression of Her-2 upregulates FoxM1 in gastric cancer. Int. J. Mol. Med. 2014, 33, 1531–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tio, D.; Kasiem, F.R.; Willemsen, M.; van Doorn, R.; van der Werf, N.; Hoekzema, R.; Luiten, R.M.; Bekkenk, M.W. Expression of cancer/testis antigens in cutaneous melanoma: A systematic review. Melanoma Res. 2019, 29, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-M.; Chang, W.-C.; Ma, W.-L. Hypothesis: Solid tumours behave as systemic metabolic dictators. J. Cell. Mol. Med. 2016, 20, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, Y.; Sang, M.; Gu, L.; Liu, F.; Yin, D.; Liu, S.; Huang, W.; Wu, Y.; Shan, B. MAGE-A family is involved in gastric cancer progression and indicates poor prognosis of gastric cancer patients. Pathol. Res. Pract. 2017, 213, 943–948. [Google Scholar] [CrossRef]
- Garus, A.; Autexier, C. Dyskerin: An essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA 2021, 27, 1441–1458. [Google Scholar] [CrossRef]
- Hermes, N.; Kewitz, S.; Staege, M.S. Preferentially Expressed Antigen in Melanoma (PRAME) and the PRAME Family of Leucine-Rich Repeat Proteins. Curr. Cancer Drug Targets 2016, 16, 400–414. [Google Scholar] [CrossRef]
- Napolitano, G.; Tagliaferri, D.; Fusco, S.; Cirillo, C.; De Martino, I.; Addeo, M.; Mazzone, P.; Russo, N.A.; Natale, F.; Cardoso, M.C.; et al. A novel member of Prame family, Gm12794c, counteracts retinoic acid differentiation through the methyltransferase activity of PRC2. Cell Death Differ. 2020, 27, 345–362. [Google Scholar] [CrossRef] [Green Version]
- Fernández, C.; Lobo Md, M.D.V.T.; Gómez-Coronado, D.; Lasunción, M.A. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp. Cell Res. 2004, 300, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The molecular functions of hepatocyte nuclear factors—In and beyond the liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Histological Type | ||
|---|---|---|---|
| Diffuse (n = 62) | Intestinal (n = 140) | ||
| Age (years, median with range) | 61.5 (53–70) | 67 (58–72) | |
| Gender | Woman | 27 (44%) | 47 (34%) |
| Man | 35 (56%) | 93 (66%) | |
| Anatomic | Antrum Distal | 31 (51%) | 50 (37%) |
| Cardia Proximal | 5 (8%) | 18 (13%) | |
| Fundus Body | 22 (36%) | 55 (40%) | |
| Gastroesophageal Junction | 3 (4.8%) | 13 (10%) | |
| Missing | 1 | 4 | |
| Anatomic JGCA (Japanese Gastric Cancer Association) | Distal | 31 (53%) | 50 (41%) |
| Proximal | 27 (47%) | 73 (59%) | |
| Missing | 4 | 17 | |
| Pathologic stage | I | 5 (8%) | 21 (15%) |
| II | 18 (31%) | 25 (18%) | |
| III | 31 (53%) | 70 (52%) | |
| IV | 5 (8%) | 21 (15%) | |
| Missing | 3 | 3 | |
| Pathologic T | T1 | 0 (0%) | 10 (7%) |
| T2 | 16 (26%) | 28 (20%) | |
| T3 | 23 (37%) | 64 (46%) | |
| T4 | 23 (37%) | 38 (27%) | |
| Missing | 0 | 33 | |
| Pathologic N | N0 | 13 (21%) | 33 (24%) |
| N1 | 18 (29%) | 34 (25%) | |
| N2 | 15 (24%) | 42 (31%) | |
| N3 | 16 (26%) | 27 (20%) | |
| Missing | 0 | 4 | |
| Pathologic M | M0 | 54 (90%) | 125 (91%) |
| M1 | 6 (10%) | 12 (9%) | |
| Missing | 2 | 3 | |
| Microsatellite status | MSS | 46 (74%) | 90 (64%) |
| MSI.L | 7 (11%) | 24 (17%) | |
| MSI.H | 9 (15%) | 26 (19%) | |
| Missing | 0 | 0 | |
| Primary therapy outcome success | Complete Response | 31 (55%) | 74 (63%) |
| Partial Response | 1 (2%) | 3 (3%) | |
| Stable Disease | 7 (12%) | 11 (9%) | |
| Progression Disease | 17 (30%) | 29 (25%) | |
| Missing | 6 | 23 | |
| Diffuse | Intestinal | |||
|---|---|---|---|---|
| N. Out of TOT | % | N. Out of TOT | % | |
| Hallmarks | 12/50 | 24% | 6/50 | 12% |
| GOs | 775/10,192 | 7.6% | 184/10,192 | 1.8% |
| Pathways | 926/5529 | 16.7% | 257/5529 | 4.6% |
| Chromosome positions | 6/299 | 2% | 9/299 | 3% |
| Motifs/miRNAs | 550/3735 | 14.7% | 6/3735 | 0.16% |
| Immunologic signature | 958/4872 | 19.6% | 79/4872 | 1.6% |
| TCGA | ARCG | GSE15459 | TCGA vs. ARCG | TCGA vs. GSE15459 | |
|---|---|---|---|---|---|
| Enriched in Diffuse | |||||
| hallmark | 12 | 8 | 6 | 7 * | 5 * |
| Go | 926 | 837 | 675 | 522 * | 301 * |
| Pathways | 775 | 942 | 925 | 566 * | 452 * |
| Motif | 550 | 1089 | 1762 | 391* | 480 * |
| Chromosome positions | 6 | 4 | 4 | 1 | 0 |
| Enriched in Intestinal | |||||
| hallmark | 6 | 7 | 6 | 5 * | 4 * |
| GO | 184 | 133 | 313 | 62 * | 82 * |
| Pathways | 257 | 400 | 912 | 184 * | 209 * |
| Motif | 6 | 6 | 44 | 0 | 2 * |
| Chromosome positions | 9 | 2 | 3 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russi, S.; Marano, L.; Laurino, S.; Calice, G.; Scala, D.; Marino, G.; Sgambato, A.; Mazzone, P.; Carbone, L.; Napolitano, G.; et al. Gene Regulatory Network Characterization of Gastric Cancer’s Histological Subtypes: Distinctive Biological and Clinically Relevant Master Regulators. Cancers 2022, 14, 4961. https://doi.org/10.3390/cancers14194961
Russi S, Marano L, Laurino S, Calice G, Scala D, Marino G, Sgambato A, Mazzone P, Carbone L, Napolitano G, et al. Gene Regulatory Network Characterization of Gastric Cancer’s Histological Subtypes: Distinctive Biological and Clinically Relevant Master Regulators. Cancers. 2022; 14(19):4961. https://doi.org/10.3390/cancers14194961
Chicago/Turabian StyleRussi, Sabino, Luigi Marano, Simona Laurino, Giovanni Calice, Dario Scala, Graziella Marino, Alessandro Sgambato, Pellegrino Mazzone, Ludovico Carbone, Giuliana Napolitano, and et al. 2022. "Gene Regulatory Network Characterization of Gastric Cancer’s Histological Subtypes: Distinctive Biological and Clinically Relevant Master Regulators" Cancers 14, no. 19: 4961. https://doi.org/10.3390/cancers14194961
APA StyleRussi, S., Marano, L., Laurino, S., Calice, G., Scala, D., Marino, G., Sgambato, A., Mazzone, P., Carbone, L., Napolitano, G., Roviello, F., Falco, G., & Zoppoli, P. (2022). Gene Regulatory Network Characterization of Gastric Cancer’s Histological Subtypes: Distinctive Biological and Clinically Relevant Master Regulators. Cancers, 14(19), 4961. https://doi.org/10.3390/cancers14194961