
Noncoding RNAs and Deep Learning Neural Network Discriminate Multi-Cancer Types



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Computational Environment and Key Schematic Workflow
2.2. Data Resources
2.3. Data Preparation
2.4. Feature Selection
2.5. Machine Learning
2.6. Final Graphing
3. Results
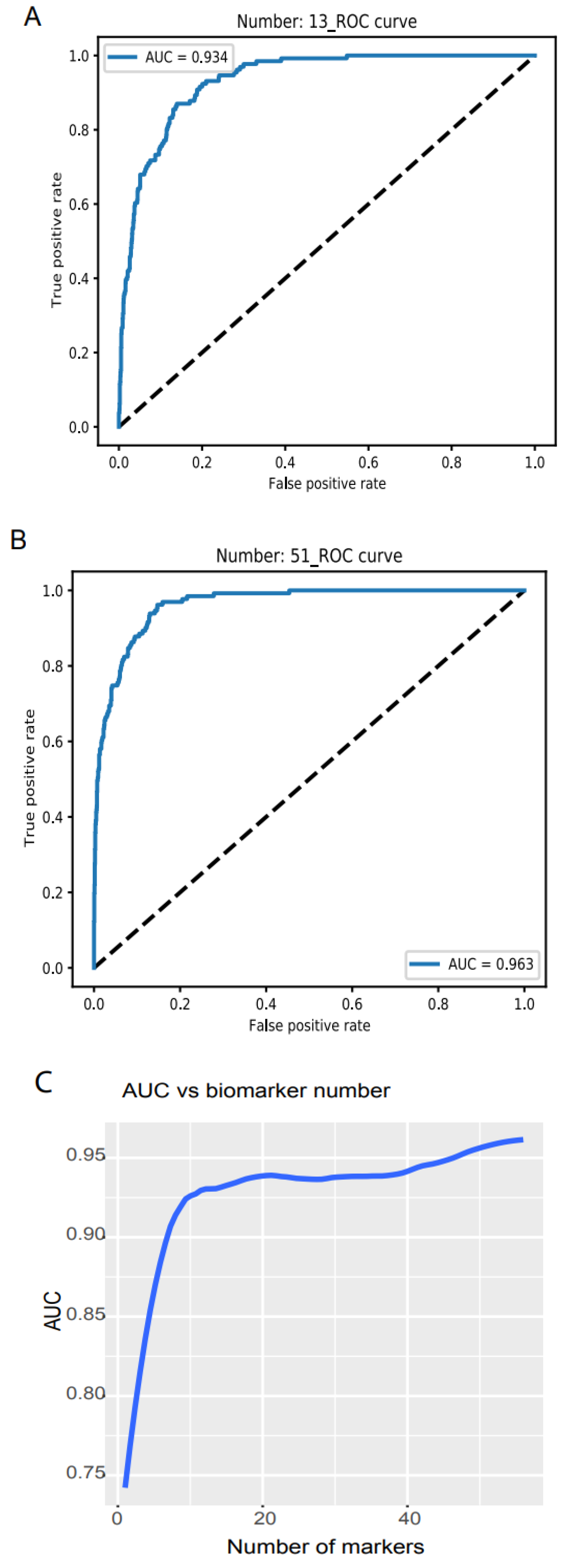
3.1. Cancer and Healthy Object Discrimination

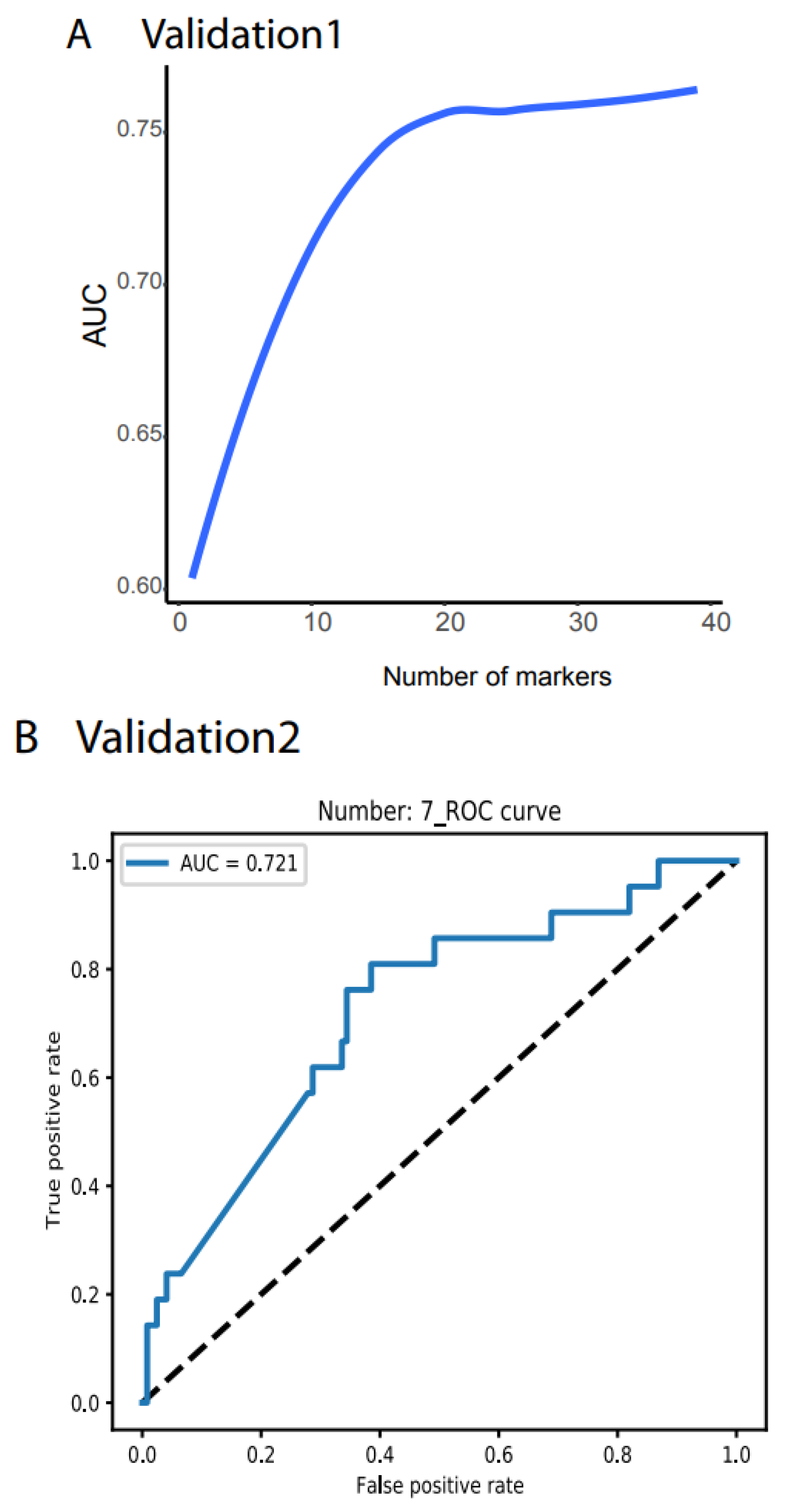
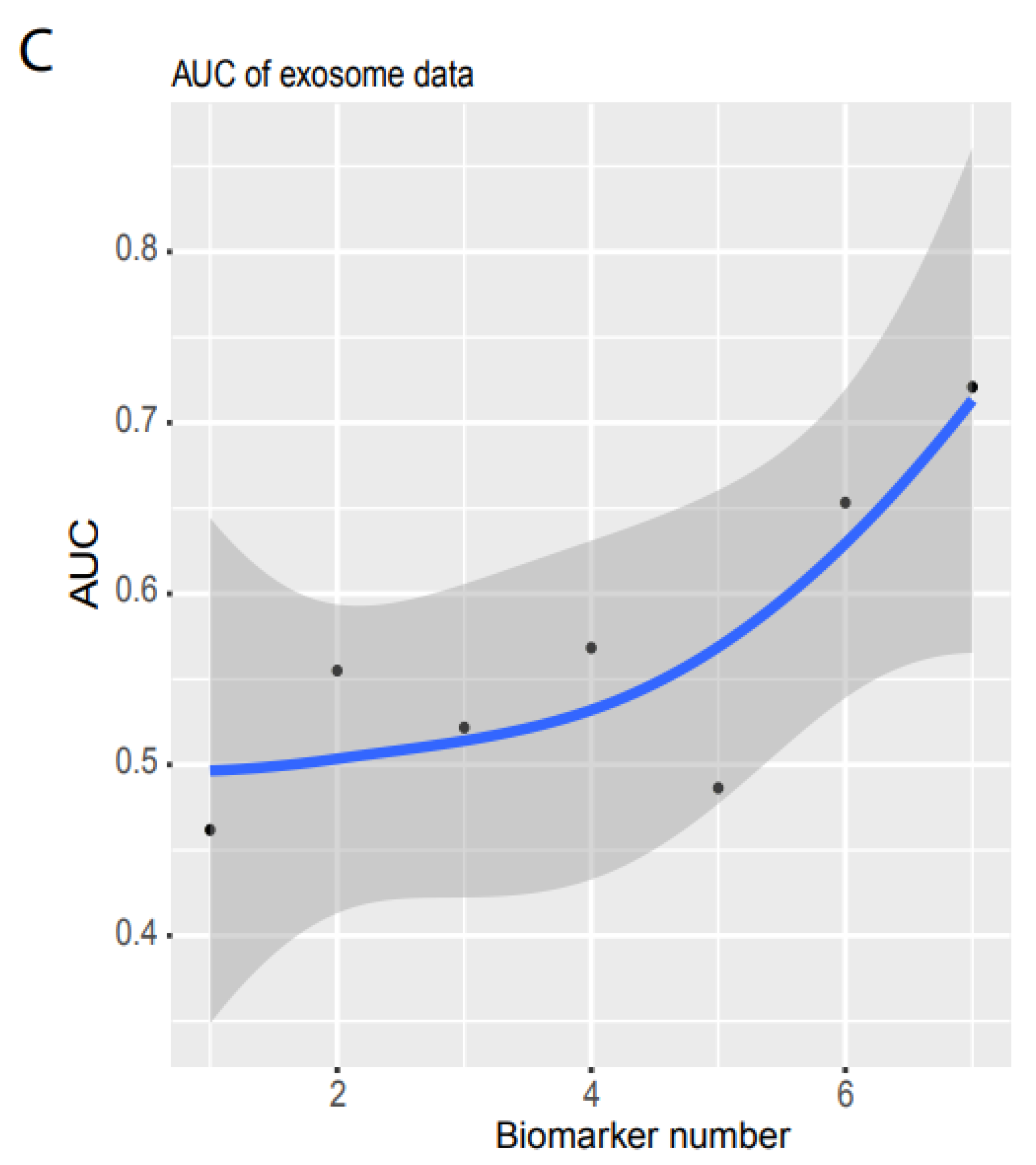
3.2. Validation
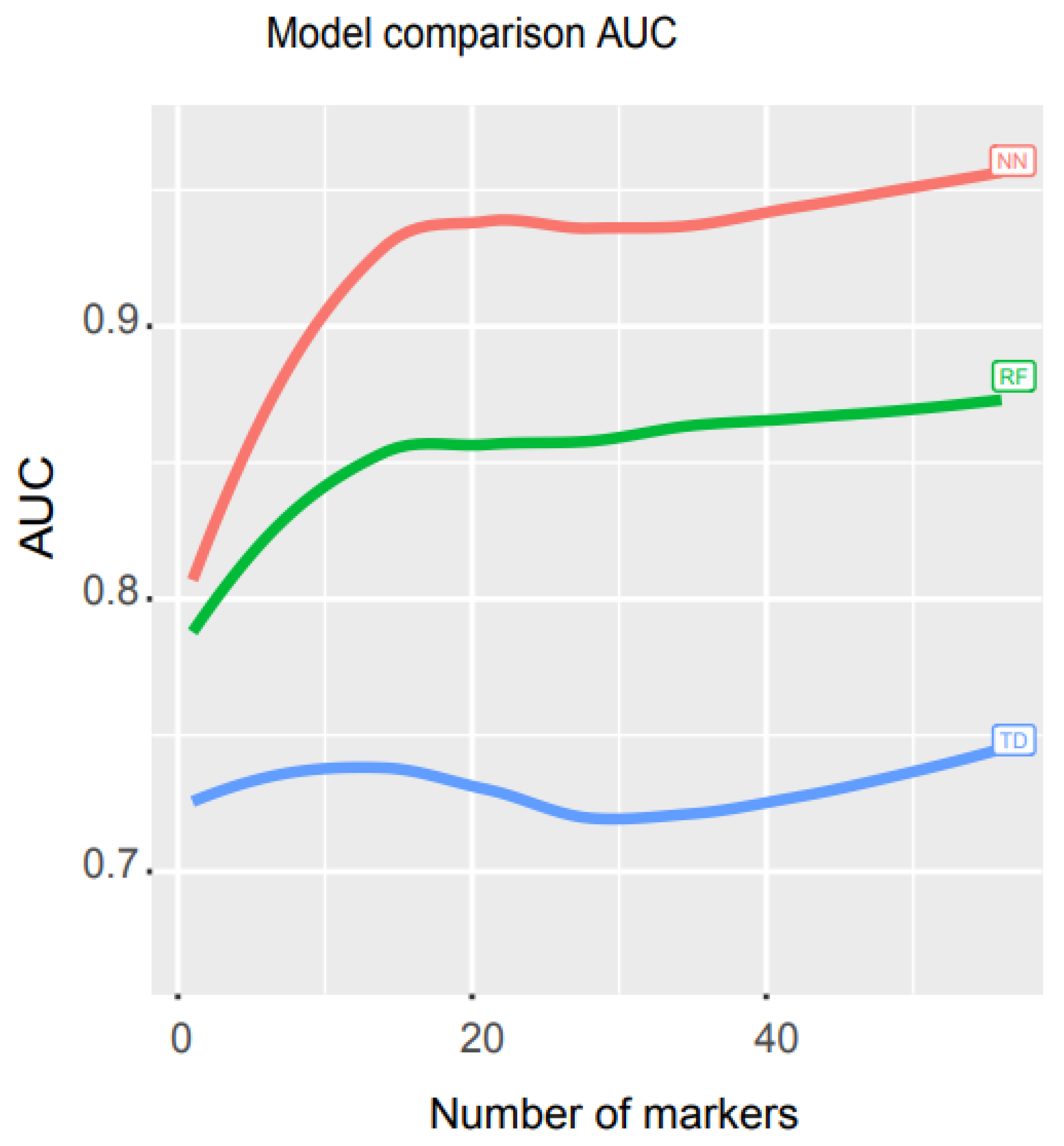
3.3. Performance Comparison of Our Model with Other Mathematical Models
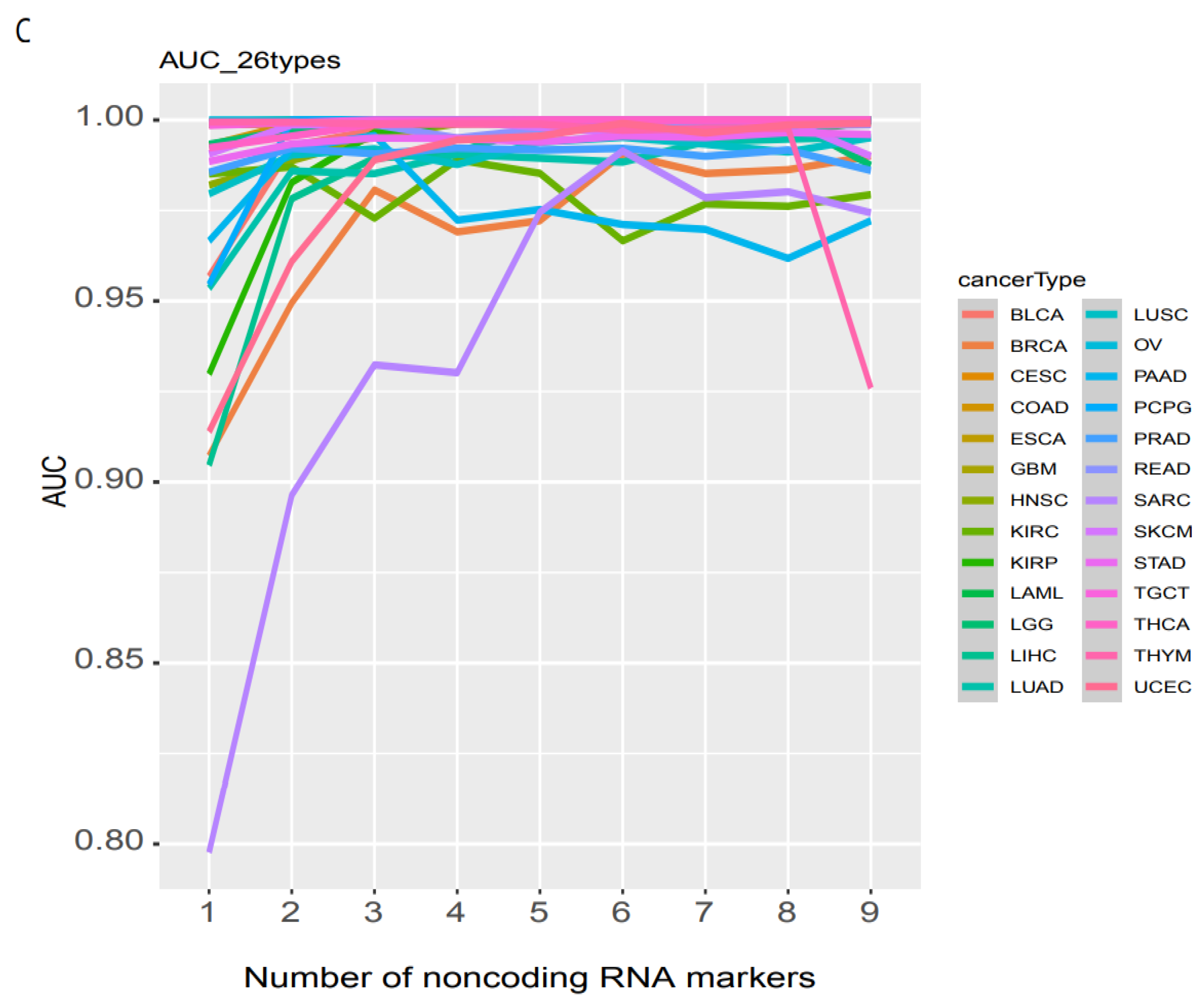
3.4. Individual Cancer Type Discrimination
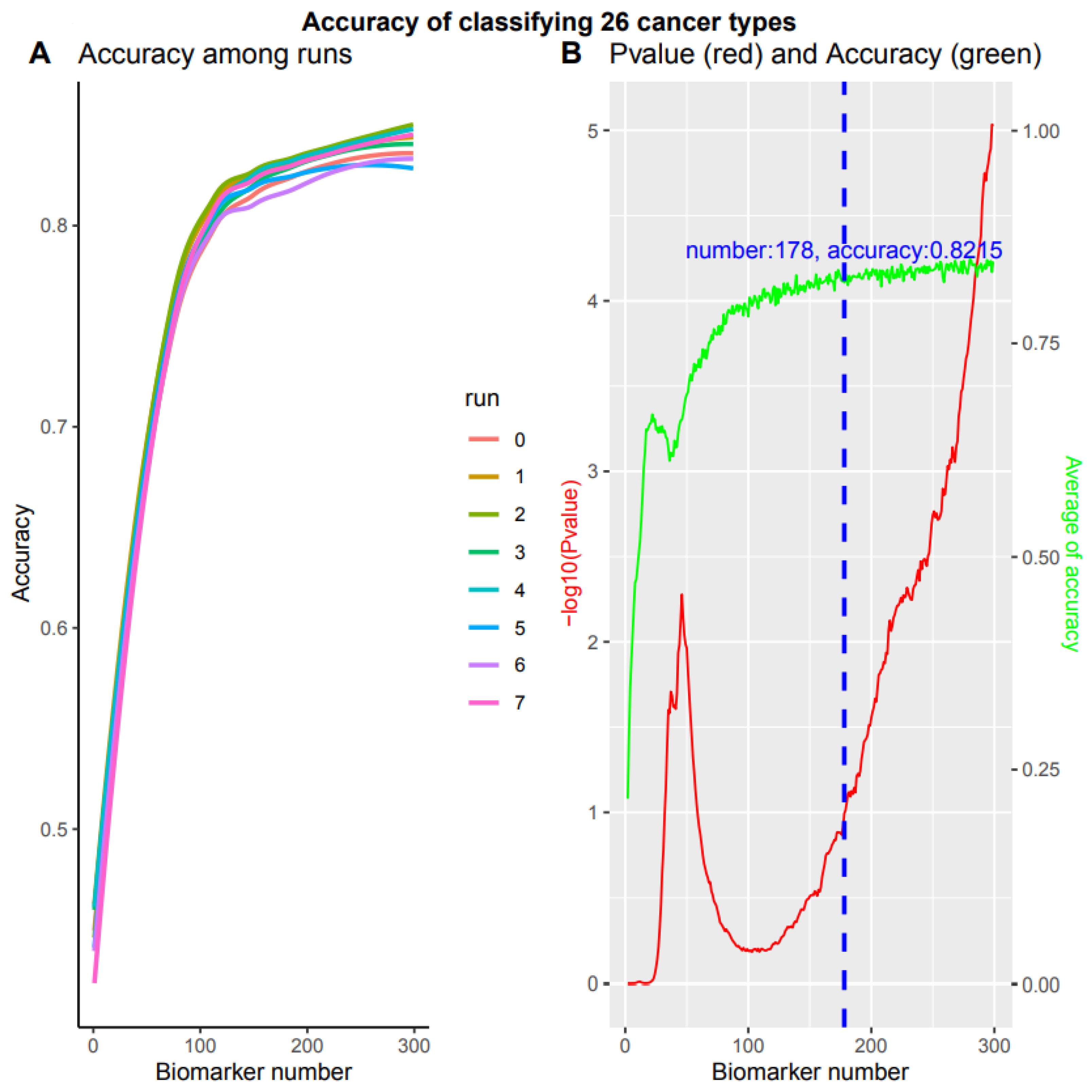
3.5. A Comprehensive Biomarker Panel for Multiple Cancer Classifiers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savage, N. How AI is improving cancer diagnostics. Nature 2020, 579, S14–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogrady, B. How cancer genomics is transforming diagnosis and treatment. Nature 2020, 579, S10–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 2020, 20, 247–257. [Google Scholar] [CrossRef]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef]
- Pashayan, N.; Pharoah, P.D.P. The challenge of early detection in cancer. Science 2020, 368, 589–590. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Brody, H. Cancer diagnosis. Nature 2020, 579, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulasingam, V.; Diamandis, E.P. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat. Clin. Pract. Oncol. 2008, 5, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Xiao, J.-F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in bladder cancer biology and therapy. Nat. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. Abstract 3287: An integrated TCGA pan-cancer clinical data resource to drive high quality survival outcome analytics. Bioinform. Syst. Biol. 2018, 78, 3287. [Google Scholar]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review the Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early stage NSCLC-challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hai, R. Noncoding RNAs Serve as the Deadliest Universal Regulators of all Cancers. Cancer Genom. Proteom. 2021, 18, 43–52. [Google Scholar] [CrossRef]
- Wang, A.; Hai, R. Noncoding RNAs Endogenously Rule the Cancerous Regulatory Realm while Proteins Govern the Normal. Available online: https://www.biorxiv.org/content/10.1101/791970v2 (accessed on 29 November 2021).
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Hu, B.; Huber, S.; Elinav, E.; Flavell, R.A. The Fire Within: Microbes Inflame Tumors. Cell 2014, 157, 776–783. [Google Scholar] [CrossRef] [Green Version]
- Howard, T.P.; Vazquez, F.; Tsherniak, A.; Hong, A.L.; Rinne, M.; Aguirre, A.J.; Boehm, J.S.; Hahn, W.C. Functional Genomic Characterization of Cancer Genomes. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Davidson, N.R.; Demircioğlu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Lehmann, K.V.; Liu, F.; Shiraishi, Y.; Soulette, C.M.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Garmire, L.; Calvisi, D.F.; Chua, M.S.; Kelley, R.K.; Chen, X. Harnessing big ‘omics’ data and AI for drug discovery in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 238–251. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakayama, K.I. Artificial intelligence in oncology. Cancer Sci. 2020, 111, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Boehm, K.M.; Khosravi, P.; Vanguri, R.; Gao, J.; Shah, S.P. Harnessing multimodal data integration to advance precision oncology. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef]
- Dlamini, Z.; Francies, F.Z.; Hull, R.; Marima, R. Artificial intelligence (AI) and big data in cancer and precision oncology. Comput. Struct. Biotechnol. J. 2020, 18, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Tran, W.T.; Sadeghi-Naini, A.; Lu, F.-I.; Gandhi, S.; Meti, N.; Brackstone, M.; Rakovitch, E.; Curpen, B. Computational Radiology in Breast Cancer Screening and Diagnosis Using Artificial Intelligence. Can. Assoc. Radiol. J. 2021, 72, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Li, Y.; Zhang, H.; Hu, J.; Liao, J.; Su, Y.; Li, Q.; Chen, B.; Li, C.; Wang, Z.; et al. exoRBase 2.0: An atlas of mRNA, lncRNA and circRNA in extracellular vesicles from human biofluids. Nucleic Acids Res. 2021, D118–D128. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hai, R. FINET: Fast Inferring NETwork. BMC Res. Notes 2020, 13, 1–6. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Cannistra, S.A. Gene-expression profiling in epithelial ovarian cancer. Nat. Clin. Pract. Oncol. 2008, 5, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Faratian, D.; Clyde, R.G.; Crawford, J.W.; Harrison, D.J. Systems pathology—Taking molecular pathology into a new dimension. Nat. Rev. Clin. Oncol. 2009, 6, 455–464. [Google Scholar] [CrossRef]
- Bidard, F.-C.; Pierga, J.-Y.; Soria, J.-C.; Thiery, J.P. Translating metastasis-related biomarkers to the clinic—Progress and pitfalls. Nat. Rev. Clin. Oncol. 2013, 10, 169–179. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Hai, R.; Rider, P.J.; He, Q. Noncoding RNAs and Deep Learning Neural Network Discriminate Multi-Cancer Types. Cancers 2022, 14, 352. https://doi.org/10.3390/cancers14020352
Wang A, Hai R, Rider PJ, He Q. Noncoding RNAs and Deep Learning Neural Network Discriminate Multi-Cancer Types. Cancers. 2022; 14(2):352. https://doi.org/10.3390/cancers14020352
Chicago/Turabian StyleWang, Anyou, Rong Hai, Paul J. Rider, and Qianchuan He. 2022. "Noncoding RNAs and Deep Learning Neural Network Discriminate Multi-Cancer Types" Cancers 14, no. 2: 352. https://doi.org/10.3390/cancers14020352
APA StyleWang, A., Hai, R., Rider, P. J., & He, Q. (2022). Noncoding RNAs and Deep Learning Neural Network Discriminate Multi-Cancer Types. Cancers, 14(2), 352. https://doi.org/10.3390/cancers14020352