
Differential Gene Expression Profiles between N-Terminal Domain and Ligand-Binding Domain Inhibitors of Androgen Receptor Reveal Ralaniten Induction of Metallothionein by a Mechanism Dependent on MTF1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
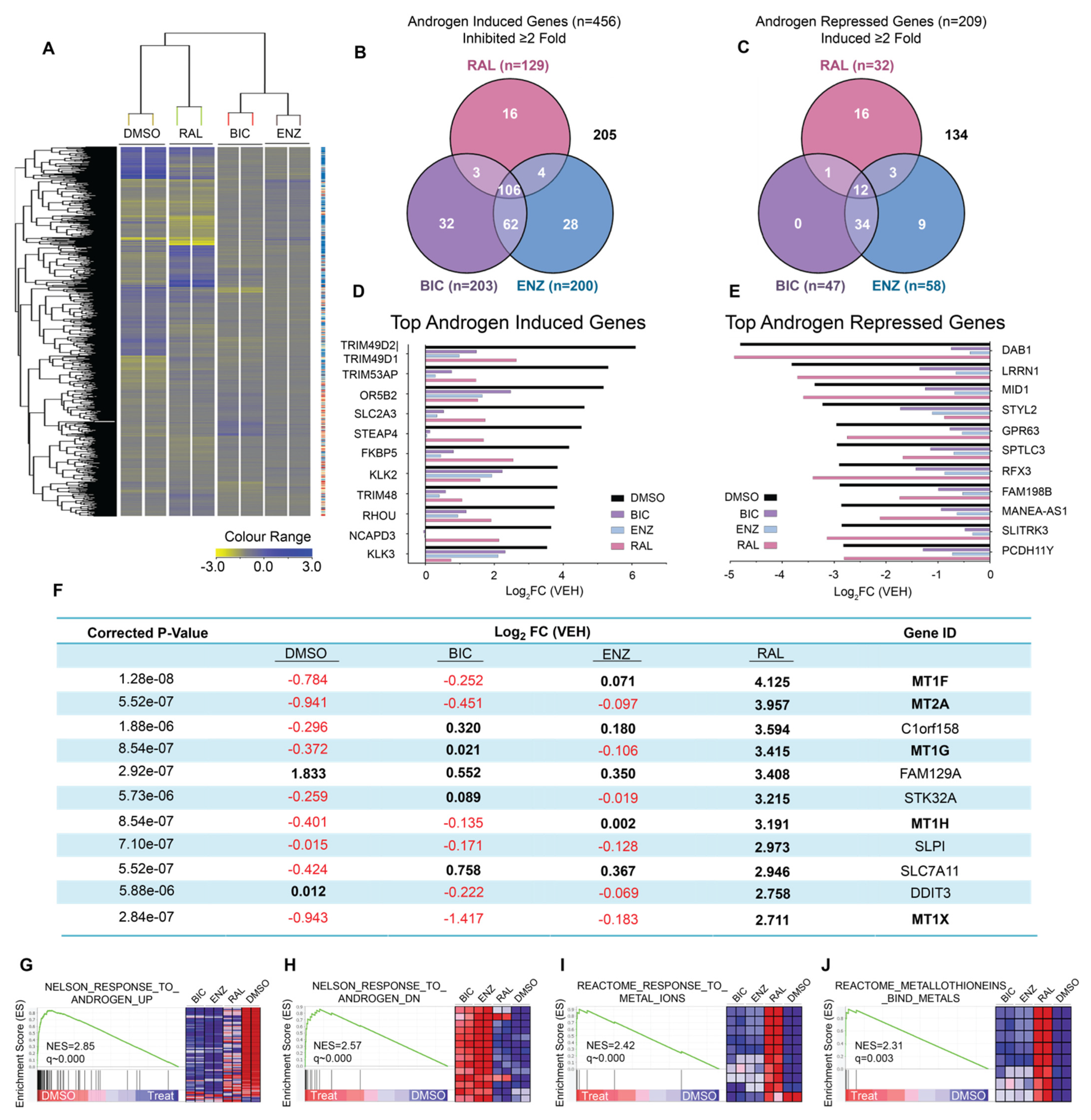
2.1. Ralaniten Has a Unique Molecular Signature
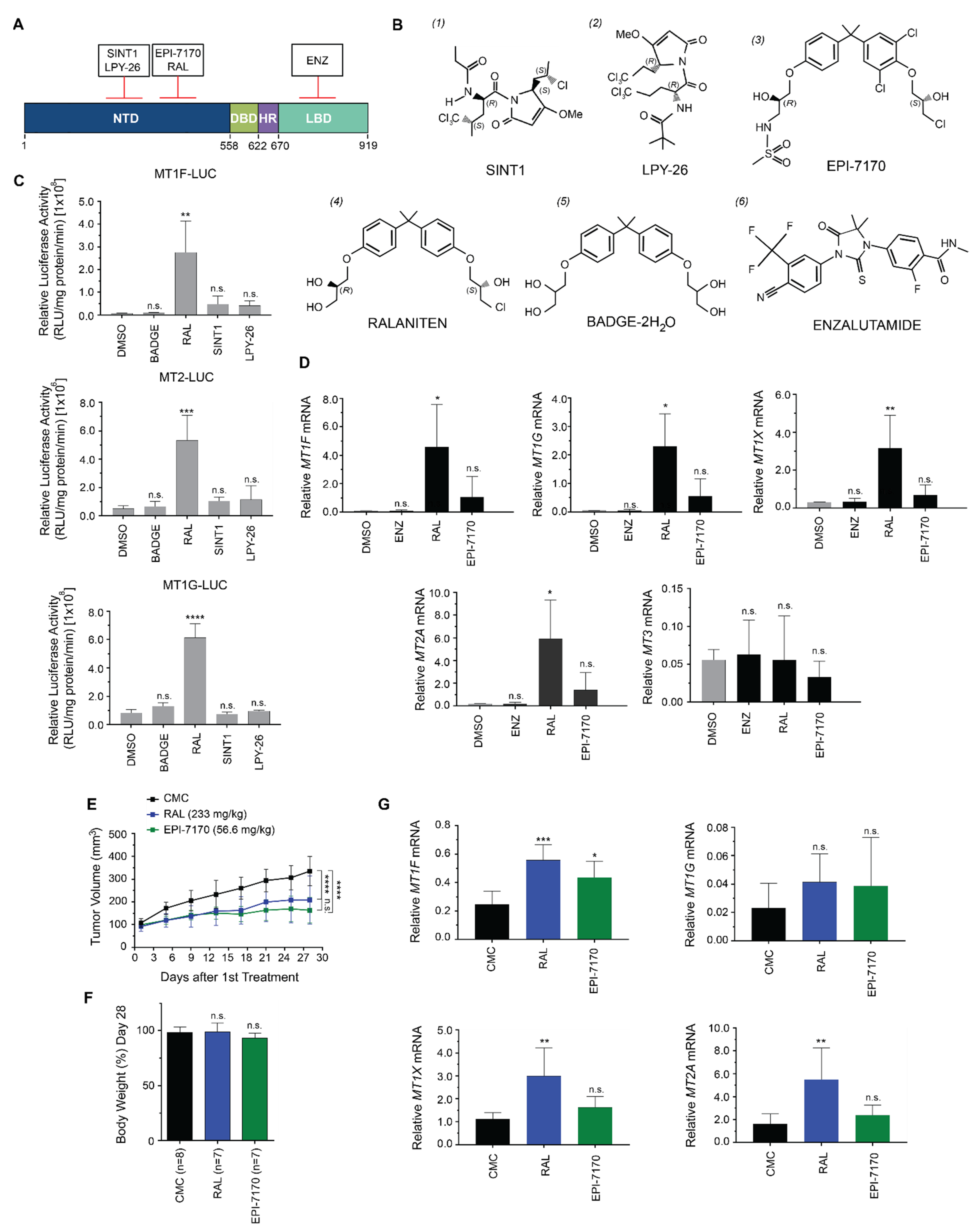
2.2. Induction of Metallothioneins Is Specific to Ralaniten
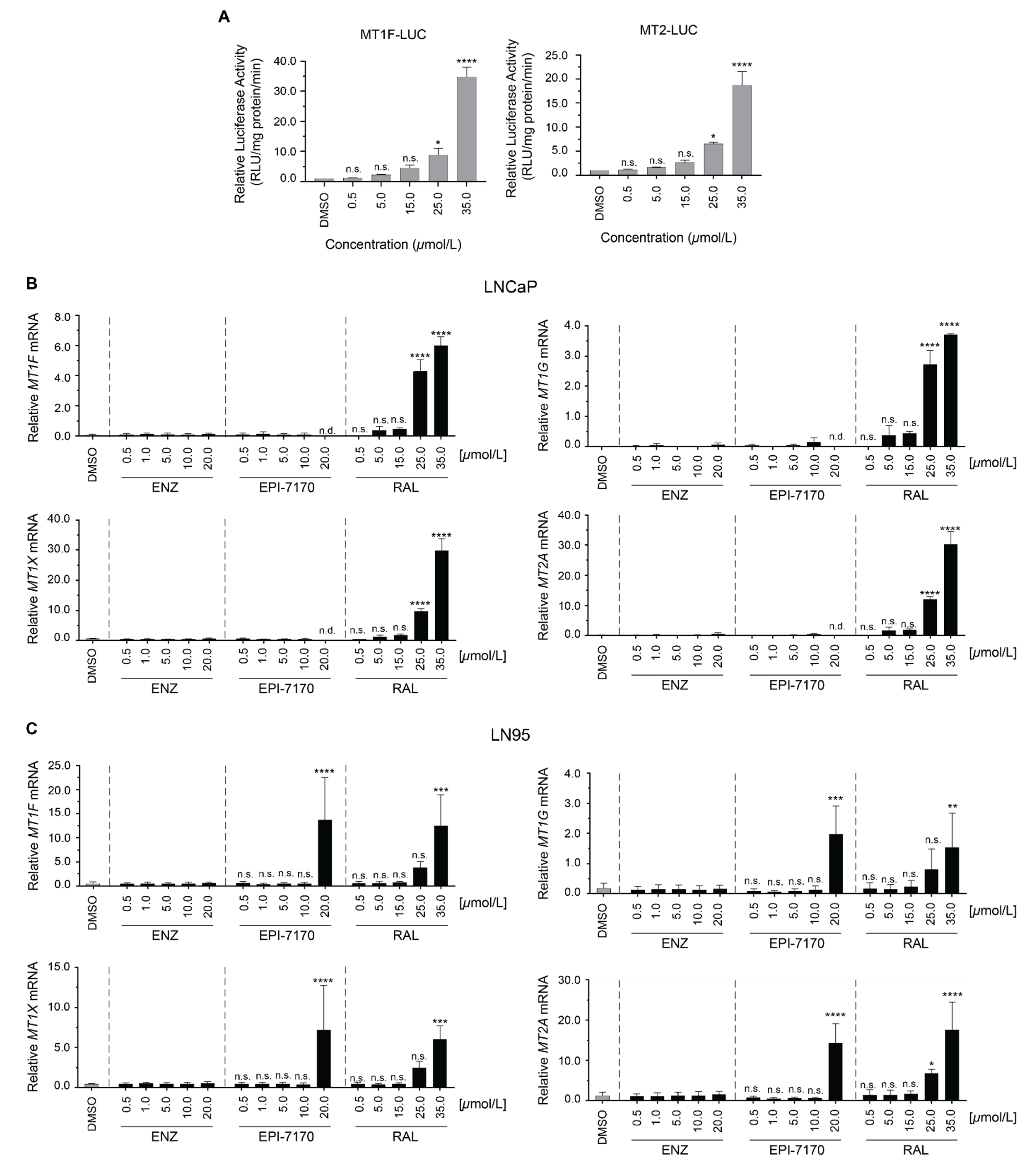
2.3. Dose-Dependent Induction of Metallothionein by Ralaniten
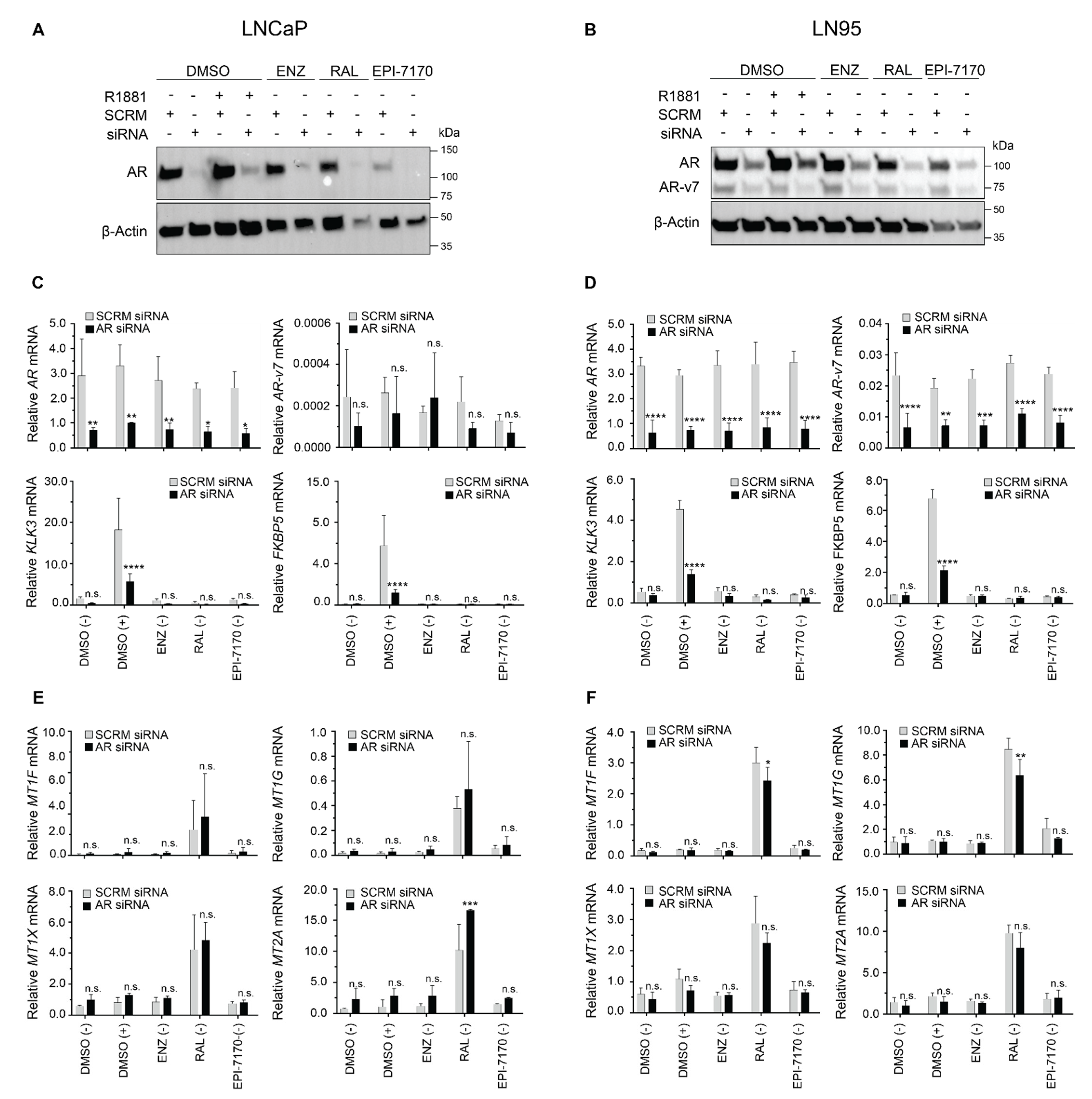
2.4. Induction of Metallothionein Isoforms by Ralaniten Is not Dependent upon the AR
2.5. Induction of Metallothionein Isoforms by Ralaniten Is Not Associated with NRF2 Expression or Increased Oxidative Stress
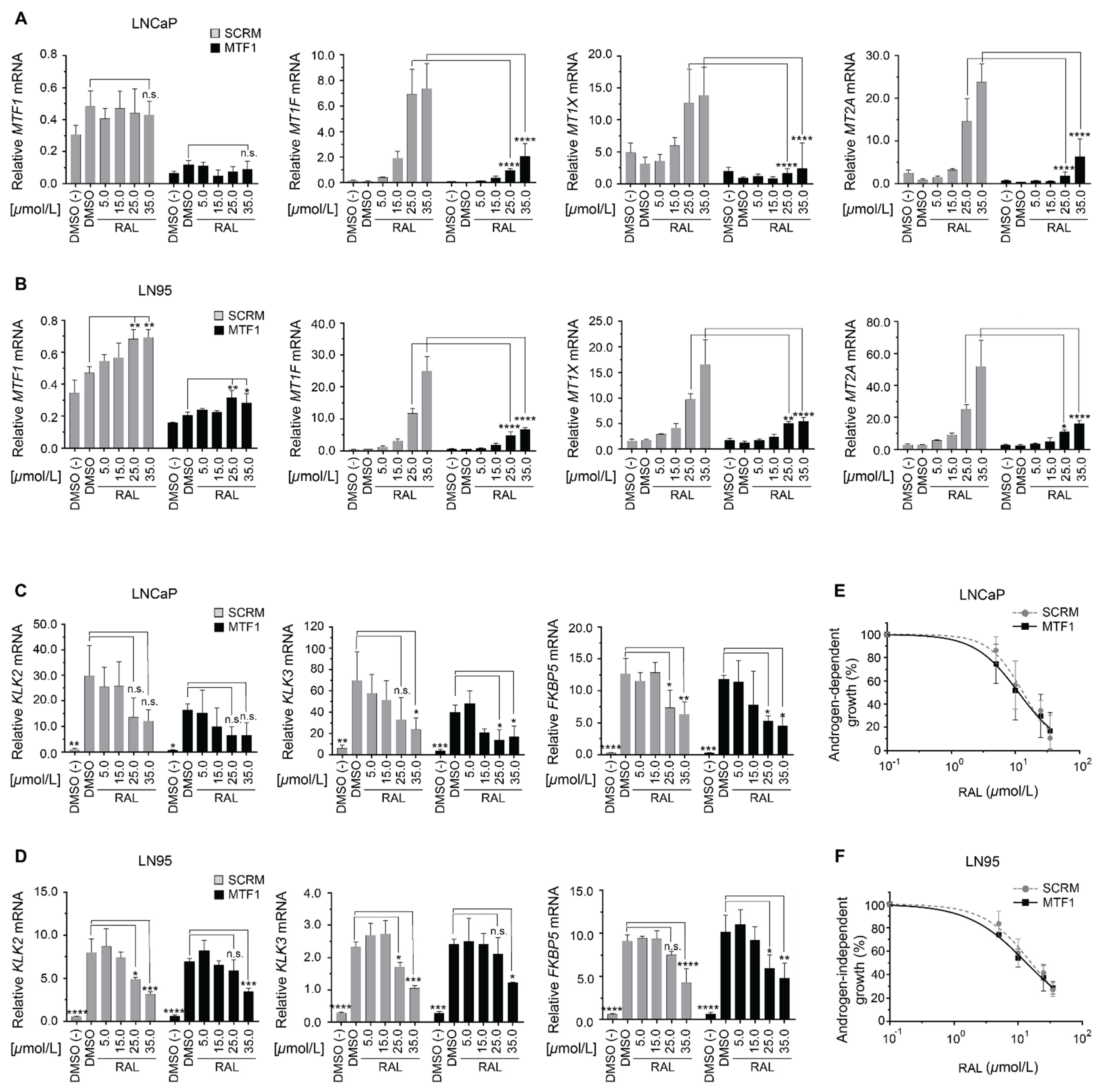
2.6. Induction of Metallothionein Isoforms by Ralaniten Is Dependent upon MTF1
3. Materials and Methods
3.1. Chemicals and Compounds
3.2. Cell Culture
3.3. Microarray and GSEA Analysis
3.4. Reporter Assays
3.5. Xenografts and Animal Experiments
3.6. Gene-Expression and Dose-Escalation Experiments
3.7. siRNA Knockdown Experiments
3.8. ROS Detection
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Sawyers, C.L. Biology of Progressive, Castration-Resistant Prostate Cancer: Directed Therapies Targeting the Androgen-Receptor Signaling Axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, F.S.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.I.; Shore, N.D.; Armstrong, A.J.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral De Novo Steroid Synthesis Activates Androgen Receptor in Castration-Resistant Prostate Cancer and Is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 165, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vassella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, S.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A Clinically Relevant Androgen Receptor Mutation Confers Resistance to Second-Generation Antiandrogens Enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Taplin, M.E.; Bubley, G.J.; Ko, Y.J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999, 59, 2511–2515. [Google Scholar]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2157. [Google Scholar] [CrossRef]
- Jenster, G.; van der Korput, H.A.; Trapman, J.; Brinkmann, A.O. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem. 1995, 270, 7341–7346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenster, G.; van der Korput, H.A.; van Vroonhoven, C.; van der Kwast, T.H.; Trapman, J.; Brinkman, A.O. Domains of the Human Androgen Receptor Involved in Steroid Binding, Transcriptional Activation, and Subcellular Localization. Mol. Endocrinol. 1991, 5, 1396–1404. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Banuelos, C.A.; Tavakoli, I.; Tien, A.H.; Caley, D.P.; Mawji, N.R.; Li, Z.; Wang, J.; Yang, Y.C.; Imamura, Y.; Yan, L.; et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. J. Biol. Chem. 2016, 291, 22231–22243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obst, J.K.; Wang, J.; Jian, K.; Williams, D.E.; Tien, A.H.; Mawji, N.R.; Tam, T.; Yang, Y.C.; Andersen, R.J.; Chi, K.N.; et al. Revealing Metabolic Liabilities of Ralaniten To Enhance Novel Androgen Receptor Targeted Therapies. ACS Pharmacol. Transl. Sci. 2019, 2, 453–467. [Google Scholar] [CrossRef]
- Hiryama, Y.; Tam, T.; Jian, K.; Andersen, R.J.; Sadar, M.D. Combination therapy with androgen receptor N-terminal domain antagonist EPI-7170 and enzalutamide yields synergistic activity in AR-V7-positive prostate cancer. Mol. Oncol. 2020, 14, 2455–2470. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, C.A.; Ito, Y.; Obst, J.K.; Mawji, N.R.; Wang, J.; Hirayama, Y.; Leung, J.K.; Tam, T.; Tien, A.H.; Andersen, R.J.; et al. Ralaniten Sensitizes Enzalutamide-Resistant Prostate Cancer to Ionizing Radiation in Prostate Cancer Cells that Express Androgen Receptor Splice Variants. Cancers 2020, 12, 1991. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.K.; Imamura, Y.; Kato, M.; Wang, J.; Mawji, N.R.; Sadar, M.D. Pin1 inhibition improves the efficacy of ralaniten compounds that bind to the N-terminal domain of androgen receptor. Commun. Biol. 2021, 4, 381. [Google Scholar] [CrossRef] [PubMed]
- Tien, A.H.; Sadar, M.D. Cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with ralaniten analogues for the treatment of androgen receptor-positive prostate and breast cancers. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Bchler, R.H.; Kagi, J.H. Human hepatic metallothioneins. FEBS Lett. 1974, 39, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Shock, H.; Demoor, J.M.; Kennette, W.A.; Collins, O.M.; Koropatnick, J. Zinc-Metallothionein Levels Are Correlated with Enhanced Glucocorticoid Responsiveness in Mouse Cells Exposed to ZnCl(2), HgCl(2), and heat shock. Toxicol. Sci. 2001, 76, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Desouki, M.M.; Lin, S.; Xiao, D.; Franklin, R.B.; Feng, P. Differential expression of metallothioneins (MTs) 1,2, and 3 in response to zinc treatment in human prostate normal and malignant cells and tissues. Mol. Cancer 2008, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, T.P.; Li, Q.; Bittel, D.; Liang, L.; Andrews, G.K. Oxidative Stress Activates Metal-responsive Transcription Factor-1 Binding Activity. Occupancy in vivo of Metal response Elements in the Metallothionein-I Gene Promoter. J. Biol. Chem. 1996, 271, 26233–26241. [Google Scholar] [CrossRef] [Green Version]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hetmatol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Bainbridge, M.N.; Warren, R.L.; Hirst, M.; Romanuik, T.; Zeng, T.; Go, A.; Delany, A.; Griffith, M.; Hickenbotham, M.; Magrini, V.; et al. Analysis of the prostate cnacer cell line LNCaP transcriptome using a sequence-by-synthesis approach. BMC Genom. 2006, 7, 246. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.S.; Clegg, N.; Arnold, H.; Ferguson, C.; Bonham, M.; White, J.; Hood, L.; Lin, B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 11890–11895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanuik, T.L.; Wang, G.; Holt, R.A.; Jones, S.J.M.; Marra, M.A.; Sadar, M.D. Indentification of novel androgen-responsive genes by sequencing of LongSAGE libraries. BMC Genom. 2009, 10, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse, A.; Bartsch, S.; Baniahmad, A. Androgen receptor-mediated gene repression. Mol. Cell. Endocrinol. 2012, 352, 46–56. [Google Scholar] [CrossRef]
- Andrews, G.K. Regulation of Metallothionein Gene Expression by Oxidative Stress and Metal Ions. Biochem. Pharmacol. 1999, 59, 95–104. [Google Scholar] [CrossRef]
- Coyle, P.; Philcox, J.C.; Carely, L.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Cleutjens, C.B.; Steketee, K.; van Eekelen, C.C.; van der Korput, J.A.; Brinkmann, A.O.; Trapman, J. Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulation properties of LNCaP cells. Endocrinology 1997, 12, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.Y.; Lu, S.X.; Xu, G.; Liu, X.D.; Li, J.; Zhang, D.S. Expression of metallothionein and Nrf2 pathway genes in lung cancer and cancer-surrounding tissues. World J. Surg. Oncol. 2013, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Lossow, K.; Kopp, J.F.; Schwerdtle, T.; Kipp, A.P. Crosstalk of Nrf2 with the Trace Elements Selenium, Iron, Zinc, and Copper. Nutrients 2019, 11, 2112. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inhereted DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistanct Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Jasani, B.; Schmid, K.W. Significance of metallothionein overexpression in human tumours. Histopathology 1997, 31, 211–214. [Google Scholar] [CrossRef]
- Han, Y.C.; Zheng, Z.L.; Zuo, Z.H.; Yu, Y.P.; Chen, R.; Tseng, G.C.; Nelson, J.B.; Luo, J.H. Metallothionein 1 h tumour suppressor activity in prostate cancer is mediated by euchromatin methyltransferase 1. J. Pathol. 2013, 230, 184–193. [Google Scholar] [CrossRef]
- Garrett, S.H.; Sens, M.A.; Shukla, D.; Flores, L.; Somji, S.; Todd, J.H.; Sens, D.A. Metallothionein Isoform 1 and 2 Gene Expression in the Human Prostate: Downregulation of MT-1X in Advanced Prostate Cancer. Prostate 2000, 43, 125–135. [Google Scholar] [CrossRef]
- Henrique, R.; Jeronimo, C.; Hoque, M.O.; Nomoto, S.; Carvalho, A.L.; Costa, V.L.; Oliveira, J.; Teixeira, M.R.; Lopes, C.; Sidransky, D. MT1G Hypermethylation Is Associated with Higher Tumor Stage in Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1274–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prueitt, R.L.; Yi, M.; Hudson, R.S.; Wallace, T.A.; Howe, T.M.; Yfantis, H.G.; Lee, D.H.; Stephens, R.M.; Liu, C.G.; Calin, G.A.; et al. Expression of MicroRNAs and Protein-Coding Genes Associated With Perineural Invasion in Prostate Cancer. Prostate 2008, 68, 1152–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obst, J.K.; Mawji, N.R.; Teskey, S.J.L.; Wang, J.; Sadar, M.D. Differential Gene Expression Profiles between N-Terminal Domain and Ligand-Binding Domain Inhibitors of Androgen Receptor Reveal Ralaniten Induction of Metallothionein by a Mechanism Dependent on MTF1. Cancers 2022, 14, 386. https://doi.org/10.3390/cancers14020386
Obst JK, Mawji NR, Teskey SJL, Wang J, Sadar MD. Differential Gene Expression Profiles between N-Terminal Domain and Ligand-Binding Domain Inhibitors of Androgen Receptor Reveal Ralaniten Induction of Metallothionein by a Mechanism Dependent on MTF1. Cancers. 2022; 14(2):386. https://doi.org/10.3390/cancers14020386
Chicago/Turabian StyleObst, Jon K., Nasrin R. Mawji, Simon J. L. Teskey, Jun Wang, and Marianne D. Sadar. 2022. "Differential Gene Expression Profiles between N-Terminal Domain and Ligand-Binding Domain Inhibitors of Androgen Receptor Reveal Ralaniten Induction of Metallothionein by a Mechanism Dependent on MTF1" Cancers 14, no. 2: 386. https://doi.org/10.3390/cancers14020386
APA StyleObst, J. K., Mawji, N. R., Teskey, S. J. L., Wang, J., & Sadar, M. D. (2022). Differential Gene Expression Profiles between N-Terminal Domain and Ligand-Binding Domain Inhibitors of Androgen Receptor Reveal Ralaniten Induction of Metallothionein by a Mechanism Dependent on MTF1. Cancers, 14(2), 386. https://doi.org/10.3390/cancers14020386