


Targeting KRAS in PDAC: A New Way to Cure It?



Abstract
:Simple Summary
Abstract
1. Introduction
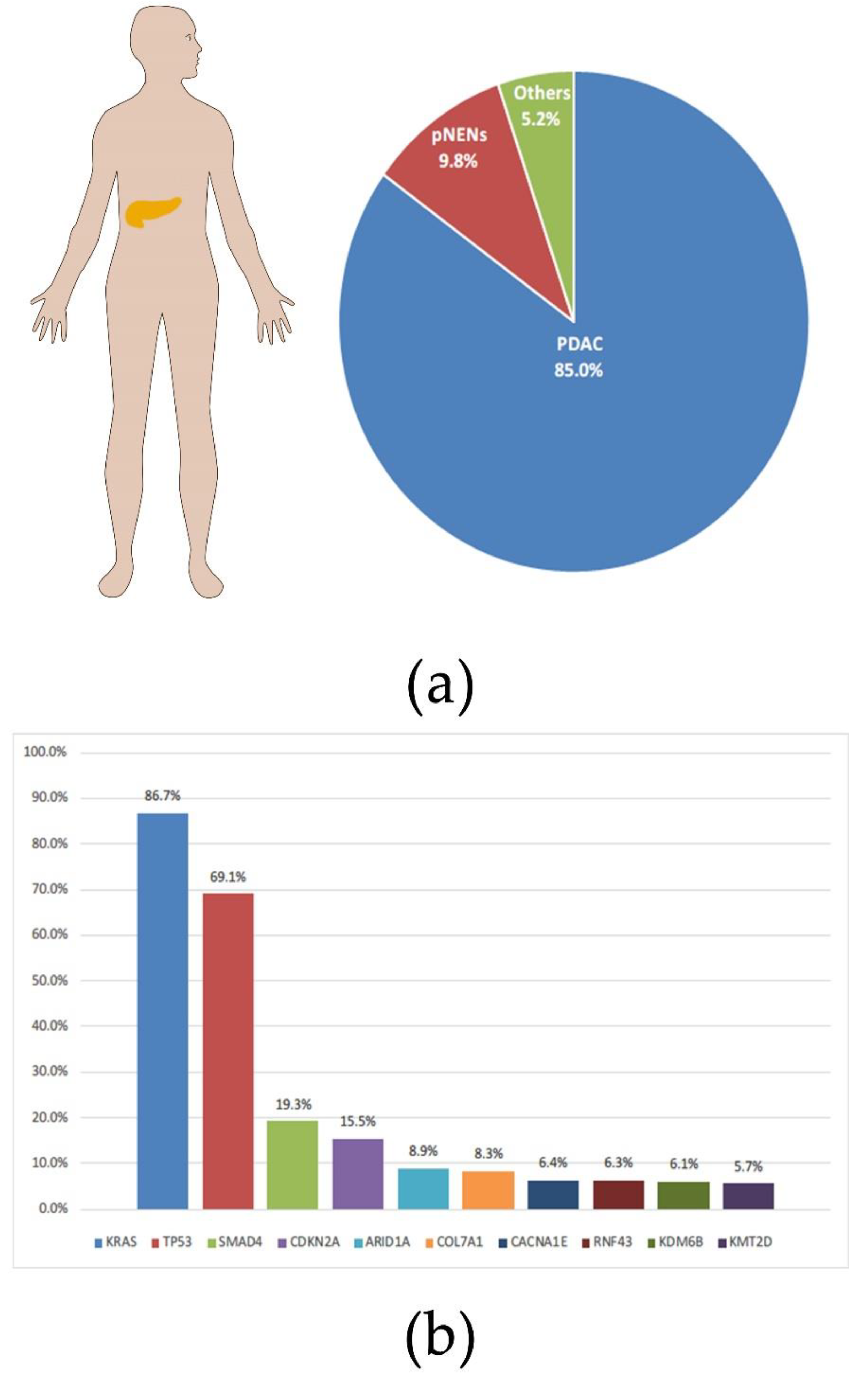
2. Clinical Status of PDAC
2.1. Living Conditions of Patients with PDAC
2.2. Clinical Treatment of PDAC
2.2.1. For Patients with Resectable Tumors
2.2.2. For Patients Who Are Unsuitable for Surgery
3. KRAS Mutations in PDAC
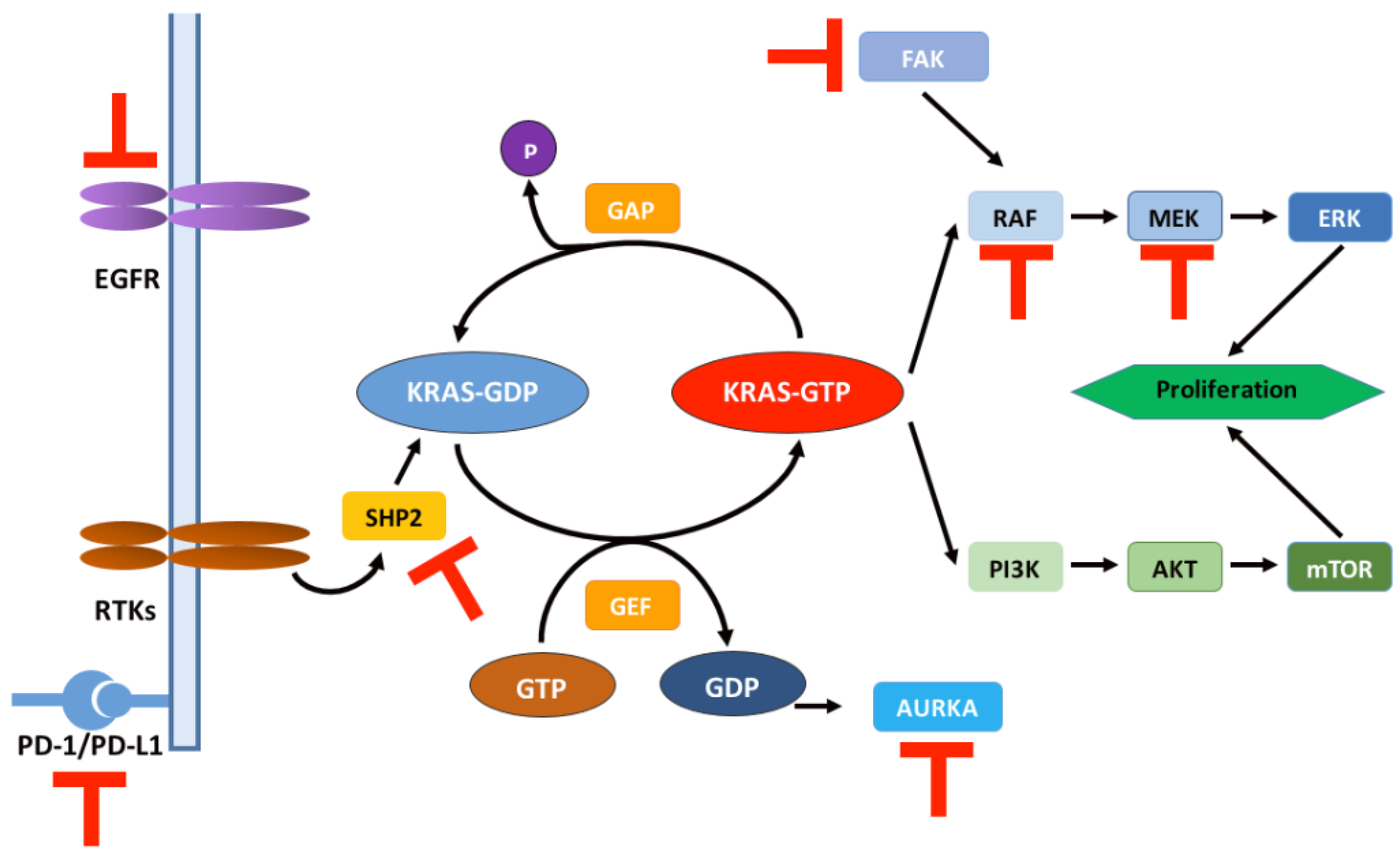
3.1. Molecular Mechanism of KRAS Mutations
3.2. Progress of PDAC with KRAS Mutations
4. KRAS Inhibitors for PDAC
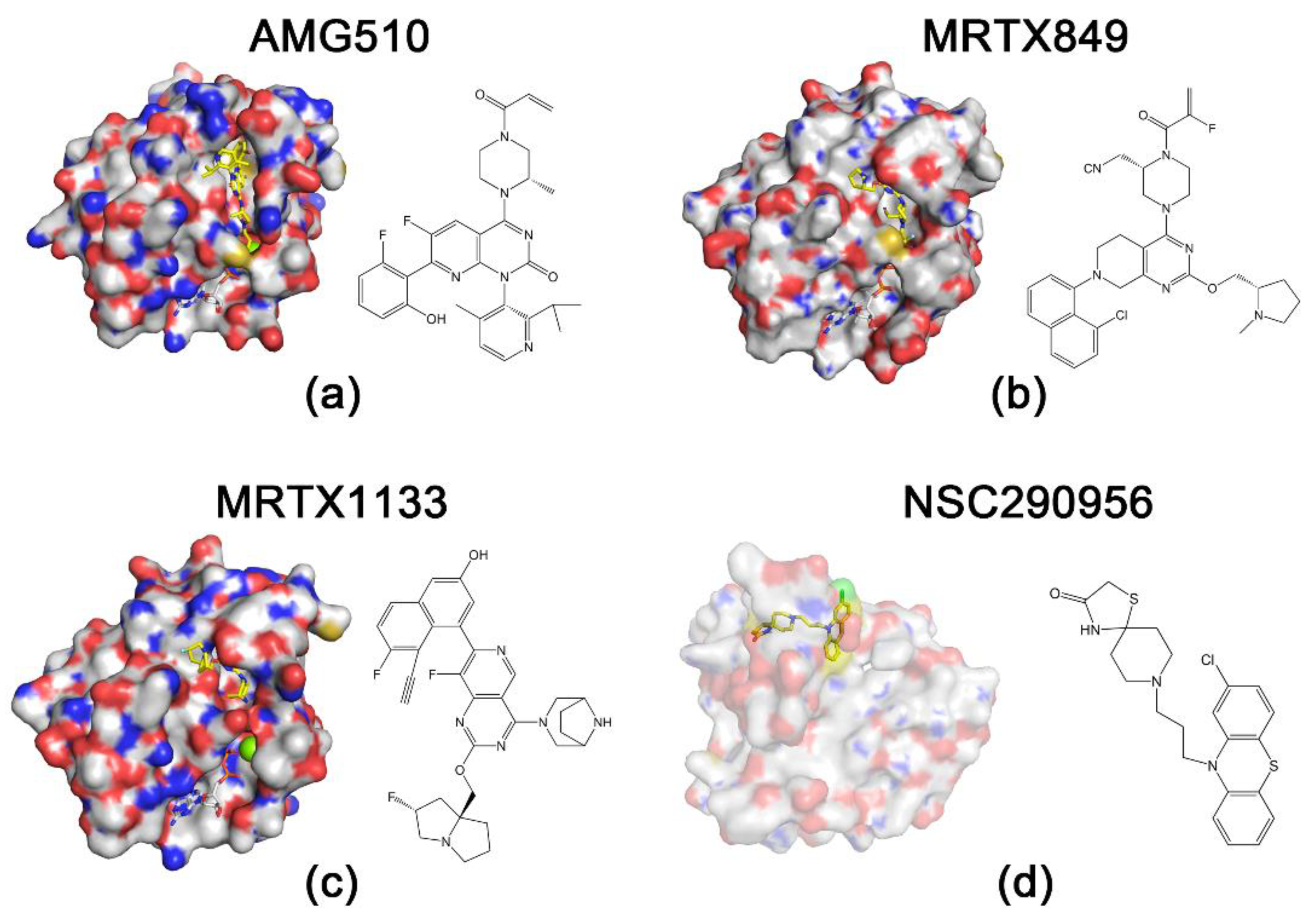
4.1. KRASG12C Inhibitors
4.2. KRASG12D Inhibitors
4.2.1. MRTX1133
4.2.2. Peptide Nucleic Acids (PNAs)
4.3. Pan-RAS Inhibitors
5. Drug Resistance Mechanisms of KRAS Inhibitors
6. Strategies to Circumvent Drug Resistance
7. Conclusions and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Ferlay, J.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Coveler, A.L. Pancreatic cancer: Optimizing treatment options, new, and emerging targeted therapies. Drug Des. Devel. Ther. 2015, 9, 3529–3545. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. Author Correction: RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 902. [Google Scholar] [CrossRef] [PubMed]
- Turpin, A.; Neuzillet, C.; Colle, E.; Dusetti, N.; Nicolle, R.; Cros, J.; de Mestier, L.; Bachet, J.B.; Hammel, P. Therapeutic advances in metastatic pancreatic cancer: A focus on targeted therapies. Ther. Adv. Med. Oncol. 2022, 14, 17588359221118019. [Google Scholar] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Zhao, W.; Yin, X.; Zheng, X.; Liu, C.; Wang, J.; Wang, E. Discovery of Small Molecule NSC290956 as a Therapeutic Agent for KRas Mutant Non-Small-Cell Lung Cancer. Front. Pharmacol. 2021, 12, 797821. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Liu, Z.; Zhao, W.; Wang, E.; Wang, J. Small Molecule APY606 Displays Extensive Antitumor Activity in Pancreatic Cancer via Impairing Ras-MAPK Signaling. PLoS ONE 2016, 11, e0155874. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, Z.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A small-molecule induces apoptosis and suppresses metastasis in pancreatic cancer cells. Eur. J. Pharm. Sci. 2013, 48, 658–667. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef]
- Chhoda, A.; Lu, L.; Clerkin, B.M.; Risch, H.; Farrell, J.J. Current Approaches to Pancreatic Cancer Screening. Am. J. Pathol. 2019, 189, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; van der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Zakelj, M.; et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: A large, international population-based study. BMC Med. 2018, 16, 125. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Millikan, K.W.; Deziel, D.J.; Silverstein, J.C.; Kanjo, T.M.; Christein, J.D.; Doolas, A.; Prinz, R.A. Prognostic factors associated with resectable adenocarcinoma of the head of the pancreas. Am. Surg. 1999, 65, 618–623. [Google Scholar] [PubMed]
- Slack, J. Developmental biology of the pancreas. Development 1995, 121, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, M.L.; Cusati, D. Total laparoscopic pancreaticoduodenectomy: Feasibility and outcome in an early experience. Arch. Surg. 2010, 145, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Palanivelu, C.; Jani, K.; Senthilnathan, P.; Parthasarathi, R.; Rajapandian, S.; Madhankumar, M.V. Laparoscopic pancreaticoduodenectomy: Technique and outcomes. J. Am. Coll. Surg. 2007, 205, 222–230. [Google Scholar] [CrossRef]
- Zureikat, A.H.; Breaux, J.A.; Steel, J.L.; Hughes, S.J. Can laparoscopic pancreaticoduodenectomy be safely implemented? J. Gastrointest. Surg. 2011, 15, 1151–1157. [Google Scholar] [CrossRef]
- Croome, K.P.; Farnell, M.B.; Que, F.G.; Reid-Lombardo, K.M.; Truty, M.J.; Nagorney, D.M.; Kendrick, M.L. Total laparoscopic pancreaticoduodenectomy for pancreatic ductal adenocarcinoma: Oncologic advantages over open approaches? Ann. Surg. 2014, 260, 633–638, discussion 638–640. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Dunn, J.A.; Stocken, D.D.; Almond, J.; Link, K.; Beger, H.; Bassi, C.; Falconi, M.; Pederzoli, P.; Dervenis, C.; et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: A randomised controlled trial. Lancet 2001, 358, 1576–1585. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs. observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs. gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy With Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Philip, P.A.; Lacy, J.; Portales, F.; Sobrero, A.; Pazo-Cid, R.; Manzano Mozo, J.L.; Kim, E.J.; Dowden, S.; Zakari, A.; Borg, C.; et al. Nab-paclitaxel plus gemcitabine in patients with locally advanced pancreatic cancer (LAPACT): A multicentre, open-label phase 2 study. Lancet Gastroenterol. Hepatol. 2020, 5, 285–294. [Google Scholar] [CrossRef]
- Lo, W.; Zureikat, A. Neoadjuvant therapy in pancreatic cancer: A review and update on recent trials. Curr. Opin. Gastroenterol. 2022, 38, 521–531. [Google Scholar] [CrossRef]
- Screening for testicular cancer:, U.S.Preventive Services Task Force reaffirmation recommendation statement. Ann. Intern. Med. 2011, 154, 483–486. [CrossRef] [Green Version]
- Zhao, Y.; Wang, Y.; Chen, W.; Bai, S.; Peng, W.; Zheng, M.; Yang, Y.; Cheng, B.; Luan, Z. Targeted intervention of eIF4A1 inhibits EMT and metastasis of pancreatic cancer cells via c-MYC/miR-9 signaling. Cancer Cell Int. 2021, 21, 670. [Google Scholar] [CrossRef]
- Marthey, L.; Sa-Cunha, A.; Blanc, J.F.; Gauthier, M.; Cueff, A.; Francois, E.; Trouilloud, I.; Malka, D.; Bachet, J.B.; Coriat, R.; et al. FOLFIRINOX for locally advanced pancreatic adenocarcinoma: Results of an AGEO multicenter prospective observational cohort. Ann. Surg. Oncol. 2015, 22, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.W.; Guo, H.; Cheng, S.; Beca, J.M.; Redmond-Misner, R.; Isaranuwatchai, W.; Qiao, L.; Earle, C.; Berry, S.R.; Biagi, J.J.; et al. Real-world outcomes of FOLFIRINOX vs. gemcitabine and nab-paclitaxel in advanced pancreatic cancer: A population-based propensity score-weighted analysis. Cancer Med. 2020, 9, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Wang, Y.; Camateros, P.; Cheung, W.Y. A Real-World Comparison of FOLFIRINOX, Gemcitabine Plus nab-Paclitaxel, and Gemcitabine in Advanced Pancreatic Cancers. J. Gastrointest. Cancer 2019, 50, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2018, PO.17.00316. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- US FDA. FDA Approves Olaparib for gBRCAm Metastatic Pancreatic Adenocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 30 December 2019).
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS(G12c) inhibitor, in advanced solid tumors. Meeting Abstract. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Drugan, J.K.; Rogers-Graham, K.; Gilmer, T.; Campbell, S.; Clark, G.J. The Ras/p120 GTPase-activating protein (GAP) interaction is regulated by the p120 GAP pleckstrin homology domain. J. Biol. Chem. 2000, 275, 35021–35027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef]
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Itonaga, M.; Ashida, R.; Murata, S.I.; Yamashita, Y.; Hatamaru, K.; Tamura, T.; Kawaji, Y.; Kayama, Y.; Emori, T.; Kawai, M.; et al. Kras Gene Analysis Using Liquid-Based Cytology Specimens Predicts Therapeutic Responses and Prognosis in Patients with Pancreatic Cancer. Cancers 2022, 14, 551. [Google Scholar] [CrossRef]
- McIntyre, C.A.; Lawrence, S.A.; Richards, A.L.; Chou, J.F.; Wong, W.; Capanu, M.; Berger, M.F.; Donoghue, M.T.A.; Yu, K.H.; Varghese, A.M.; et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer 2020, 126, 3939–3949. [Google Scholar] [CrossRef]
- Dong, L.; Wang, S.; Fu, B.; Wang, J. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci. Rep. 2018, 8, 9650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Nauheim, D.; Moskal, D.; Renslo, B.; Chadwick, M.; Jiang, W.; Yeo, C.J.; Nevler, A.; Bowne, W.; Lavu, H. KRAS mutation allele frequency threshold alters prognosis in right-sided resected pancreatic cancer. J. Surg. Oncol. 2022, 126, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Dopico, P.J.; Le, M.N.; Burgess, B.; Yang, Z.; Zhao, Y.; Wang, Y.; George, T.J.; Fan, Z.H. Longitudinal Study of Circulating Biomarkers in Patients with Resectable Pancreatic Ductal Adenocarcinoma. Biosensors 2022, 12, 206. [Google Scholar] [CrossRef]
- Li, Z.; Zhuang, H.; Chen, X.; Zhang, Y.; Ma, Z.; Wang, S.; Yan, Q.; Zhou, Z.; Huang, S.; Zhang, C.; et al. Identification of MBOAT2 as an Unfavorable Biomarker Correlated with KRAS Activation and Reduced CD8(+) T-Cell Infiltration in Pancreatic Cancer. J. Oncol. 2022, 2022, 4269733. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Brauswetter, D.; Gurbi, B.; Varga, A.; Varkondi, E.; Schwab, R.; Banhegyi, G.; Fabian, O.; Keri, G.; Valyi-Nagy, I.; Petak, I. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLoS ONE 2017, 12, e0185687. [Google Scholar] [CrossRef]
- Zhou, L.; Baba, Y.; Kitano, Y.; Miyake, K.; Zhang, X.; Yamamura, K.; Kosumi, K.; Kaida, T.; Arima, K.; Taki, K.; et al. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: Pyrosequencing technology and literature review. Med. Oncol. 2016, 33, 32. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat. Struct. Mol. Biol. 2018, 25, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Dean, D.A. Peptide nucleic acids: Versatile tools for gene therapy strategies. Adv. Drug Deliv. Rev. 2000, 44, 81–95. [Google Scholar] [CrossRef] [Green Version]
- De Cola, C.; Manicardi, A.; Corradini, R.; Izzo, I.; De Riccardis, F. Carboxyalkyl peptoid PNAs: Synthesis and hybridization properties. Article. Tetrahedron 2012, 68, 499–506. [Google Scholar] [CrossRef]
- Chiarantini, L.; Cerasi, A.; Fraternale, A.; Millo, E.; Benatti, U.; Sparnacci, K.; Laus, M.; Ballestri, M.; Tondelli, L. Comparison of novel delivery systems for antisense peptide nucleic acids. J. Control. Release 2005, 109, 24–36. [Google Scholar] [CrossRef]
- Shai, A.; Galouk, E.; Miari, R.; Tareef, H.; Sammar, M.; Zeidan, M.; Rayan, A.; Falah, M. Inhibiting mutant KRAS G12D gene expression using novel peptide nucleic acid-based antisense: A potential new drug candidate for pancreatic cancer. Oncol. Lett. 2022, 23, 130. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.B.; Ward, A.; Keeton, A.B.; Chen, X.; Maxuitenko, Y.; Prakash, A.; Li, F.; Foote, J.B.; Buchsbaum, D.J.; Piazza, G.A. Pan-RAS inhibitors: Hitting multiple RAS isozymes with one stone. Adv. Cancer Res. 2022, 153, 131–168. [Google Scholar] [PubMed]
- Keeton, A.B.; Ward, A.; Chen, X.; Valiyaveettil, J.; Zhu, B.; Ramirez-Alcantara, V. Abstract 2707: A novel RAS inhibitor, MCI-062, inhibits colon tumor growth in vivo and activates antitumor immunity. Cancer Res. 2019, 79, 2707. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [Green Version]
- Rognan, D. Rational design of protein–protein interaction inhibitors. Med. Chem. Commun. 2015, 6, 51–60. [Google Scholar] [CrossRef]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Z.; Li, D.; Wang, E.; Wang, J. Rational drug design: The search for Ras protein hydrolysis intermediate conformation inhibitors with both affinity and specificity. Curr. Pharm. Des. 2013, 19, 2246–2258. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, W.; Liu, Z.; Wang, J. Spiclomazine displays a preferential anti-tumor activity in mutant KRas-driven pancreatic cancer. Oncotarget 2018, 9, 6938–6951. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, D.; Liu, Z.; Zheng, X.; Wang, J.; Wang, E. Spiclomazine induces apoptosis associated with the suppression of cell viability, migration and invasion in pancreatic carcinoma cells. PLoS ONE 2013, 8, e66362. [Google Scholar] [CrossRef]
- Liu, Z.; Li, D.; Zheng, X.; Wang, E.; Wang, J. Selective induction of apoptosis: Promising therapy in pancreatic cancer. Curr. Pharm. Des. 2013, 19, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, D.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A potent lead induces apoptosis in pancreatic cancer cells. PLoS ONE 2012, 7, e37841. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Boned Del Rio, I.; Young, L.C.; Sari, S.; Jones, G.G.; Ringham-Terry, B.; Hartig, N.; Rejnowicz, E.; Lei, W.; Bhamra, A.; Surinova, S.; et al. SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc. Natl. Acad. Sci. USA 2019, 116, 13330–13339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2019, 179, 1239. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Zhang, M.; Song, J.; Zheng, X.; Xu, G.; Bao, Y.; Lan, J.; Luo, D.; Hu, J.; Li, J.J.; et al. Integrin-Src-YAP1 signaling mediates the melanoma acquired resistance to MAPK and PI3K/mTOR dual targeted therapy. Mol. Biomed. 2020, 1, 12. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumenschein, G.R., Jr.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Ou, S.H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. Meeting Abstract. J. Clin. Oncol. 2019, 37, 1. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [Green Version]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, C.; Velazquez, R.; Wang, H.; Dunkl, L.M.; Kazic-Legueux, M.; Haberkorn, A.; Billy, E.; Manchado, E.; Brachmann, S.M.; et al. SHP2 Inhibition Overcomes RTK-Mediated Pathway Reactivation in KRAS-Mutant Tumors Treated with MEK Inhibitors. Mol. Cancer Ther. 2019, 18, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell. Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Hata, A.N.; Shaw, A.T. Resistance looms for KRAS(G12C) inhibitors. Nat. Med. 2020, 26, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Petrenko, O.; Hayman, M.J. A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget 2015, 6, 15814–15827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jane, E.P.; Premkumar, D.R.; Addo-Yobo, S.O.; Pollack, I.F. Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J. Pharmacol. Exp. Ther. 2009, 331, 327–337. [Google Scholar] [CrossRef]
- Ozaki, K.; Kosugi, M.; Baba, N.; Fujio, K.; Sakamoto, T.; Kimura, S.; Tanimura, S.; Kohno, M. Blockade of the ERK or PI3K-Akt signaling pathway enhances the cytotoxicity of histone deacetylase inhibitors in tumor cells resistant to gefitinib or imatinib. Biochem. Biophys. Res. Commun. 2010, 391, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef] [Green Version]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.Y.; Nagasaka, M.; Li, Y.; Aboukameel, A.; Uddin, M.H.; Sexton, R.; Bannoura, S.; Mzannar, Y.; Al-Hallak, M.N.; Kim, S.; et al. Inhibitor of the Nuclear Transport Protein XPO1 Enhances the Anticancer Efficacy of KRAS G12C Inhibitors in Preclinical Models of KRAS G12C-Mutant Cancers. Cancer Res. Commun. 2022, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1-from biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. First-in-class, orally bioavailable KRAS(G12V)(ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRAS(G12V) mutant cancers. Meeting Abstract. Cancer Res. 2021, 81, 2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ClinicalTrials.Gov Identifier | Title | Phase | Drugs | Targets |
|---|---|---|---|---|
| NCT05374538 | VIC-1911 Monotherapy in Combination With Sotorasib for the Treatment of KRAS G12C-Mutant Non-Small Cell Lung Cancer | 1 | Sotorasib VIC-1911 | KRASG12C Aurora Kinase A |
| NCT05067283 | A Study of MK-1084 as Monotherapy and in Combination With Pembrolizumab (MK-3475) in Participants With KRASG12C Mutant Advanced Solid Tumors (MK-1084-001) | 1 | MK-1084 Pembrolizumab | KRASG12C PD-1 |
| NCT05379946 | Study to Evaluate D-1553 in Combination With IN10018 in Subjects With Solid Tumors | 1/2 | D-1553 IN10018 | KRASG12C FAK |
| NCT05074810 | Phase 1/2 Study of VS-6766 + Sotorasib in G12C NSCLC Patients (RAMP203) | 1/2 | Sotorasib VS-6766 | KRASG12C RAF/MEK |
| NCT05054725 | Combination Study of RMC-4630 and Sotorasib for NSCLC Subjects With KRASG12C Mutation After Failure of Prior Standard Therapies | 2 | Sotorasib RMC-4630 | KRASG12C SHP2 |
| NCT05313009 | Tarlox and Sotorasib in Patients With KRAS G12C Mutations | 1/2 | Sotorasib Tarloxotinib | KRASG12C EGFR/HER2/HER3 |
| NCT05198934 | Sotorasib and Panitumumab Versus Investigator’s Choice for Participants With Kirsten Rat Sarcoma (KRAS) p.G12C Mutation (CodeBreak 300) | 3 | Sotorasib Panitumumab | KRASG12C EGFR |
| NCT04613596 | Phase 2 Trial of MRTX849 Monotherapy and in Combination With Pembrolizumab for NSCLC With KRAS G12C Mutation KRYSTAL-7 | 2 | MRTX849 Pembrolizumab | KRASG12C PD-1 |
| NCT04330664 | Adagrasib in Combination With TNO155 in Patients With Cancer (KRYSTAL 2) | 1/2 | MRTX849 TNO155 | KRASG12C SHP2 |
| NCT05375994 | Study of VS-6766 + Adagrasib in KRAS G12C NSCLC Patients (RAMP204) | 1/2 | MRTX849 VS-6766 | KRASG12C RAF/MEK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Liu, Z.; Wang, J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers 2022, 14, 4982. https://doi.org/10.3390/cancers14204982
He Q, Liu Z, Wang J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers. 2022; 14(20):4982. https://doi.org/10.3390/cancers14204982
Chicago/Turabian StyleHe, Qianyu, Zuojia Liu, and Jin Wang. 2022. "Targeting KRAS in PDAC: A New Way to Cure It?" Cancers 14, no. 20: 4982. https://doi.org/10.3390/cancers14204982
APA StyleHe, Q., Liu, Z., & Wang, J. (2022). Targeting KRAS in PDAC: A New Way to Cure It? Cancers, 14(20), 4982. https://doi.org/10.3390/cancers14204982