
Application of CRISPR for In Vivo Mouse Cancer Studies


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Discovery of CRISPR
1.2. The Elements of CRISPR/Cas9
1.3. In Vitro Application of CRISPR
1.4. Genome Wide CRISPR Screens
1.5. In Vivo Application of CRISPR for Cancer Research
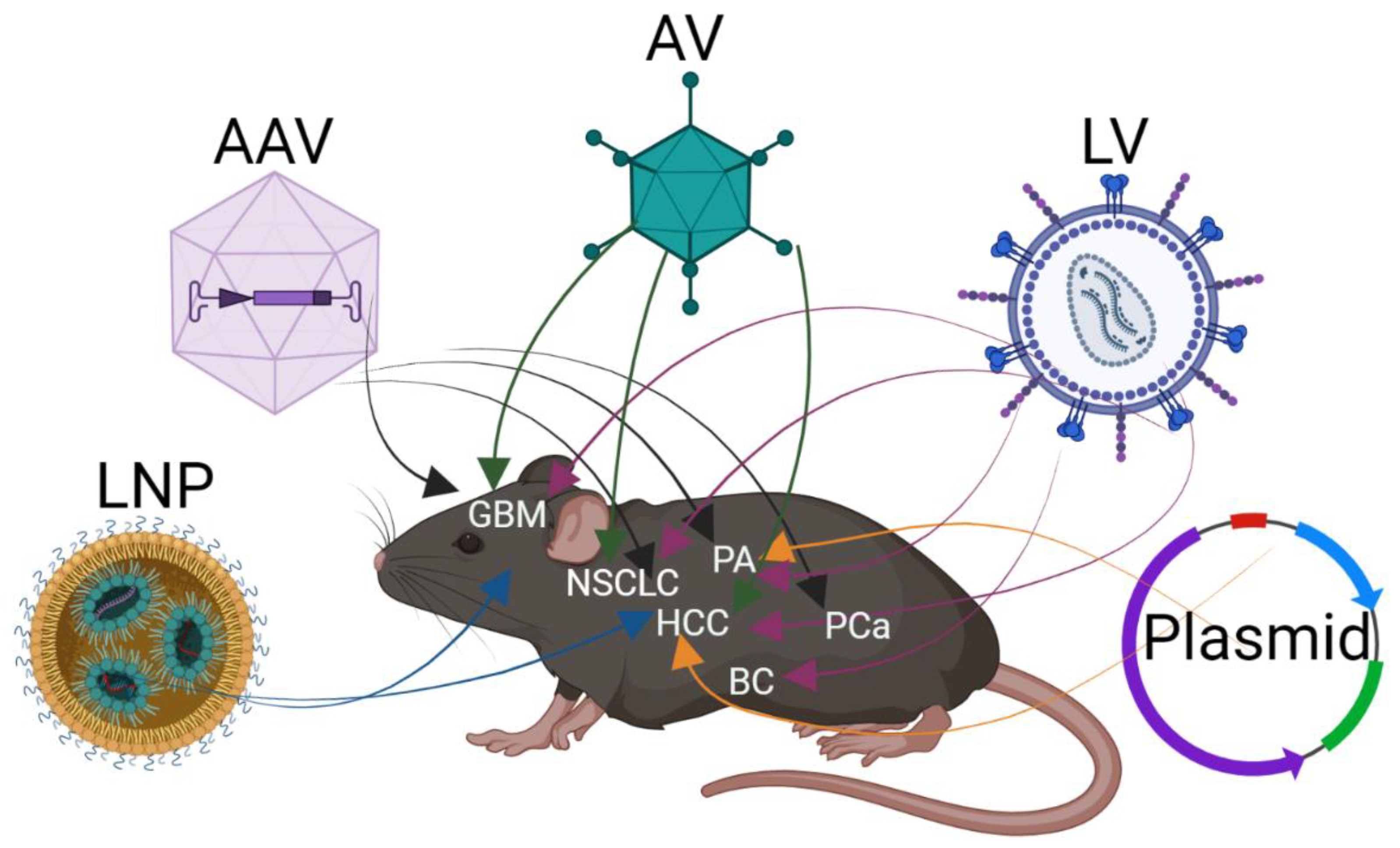
2. Delivery of CRISPR/Cas9 In Vivo to Somatic Cells
2.1. Delivery by Vector
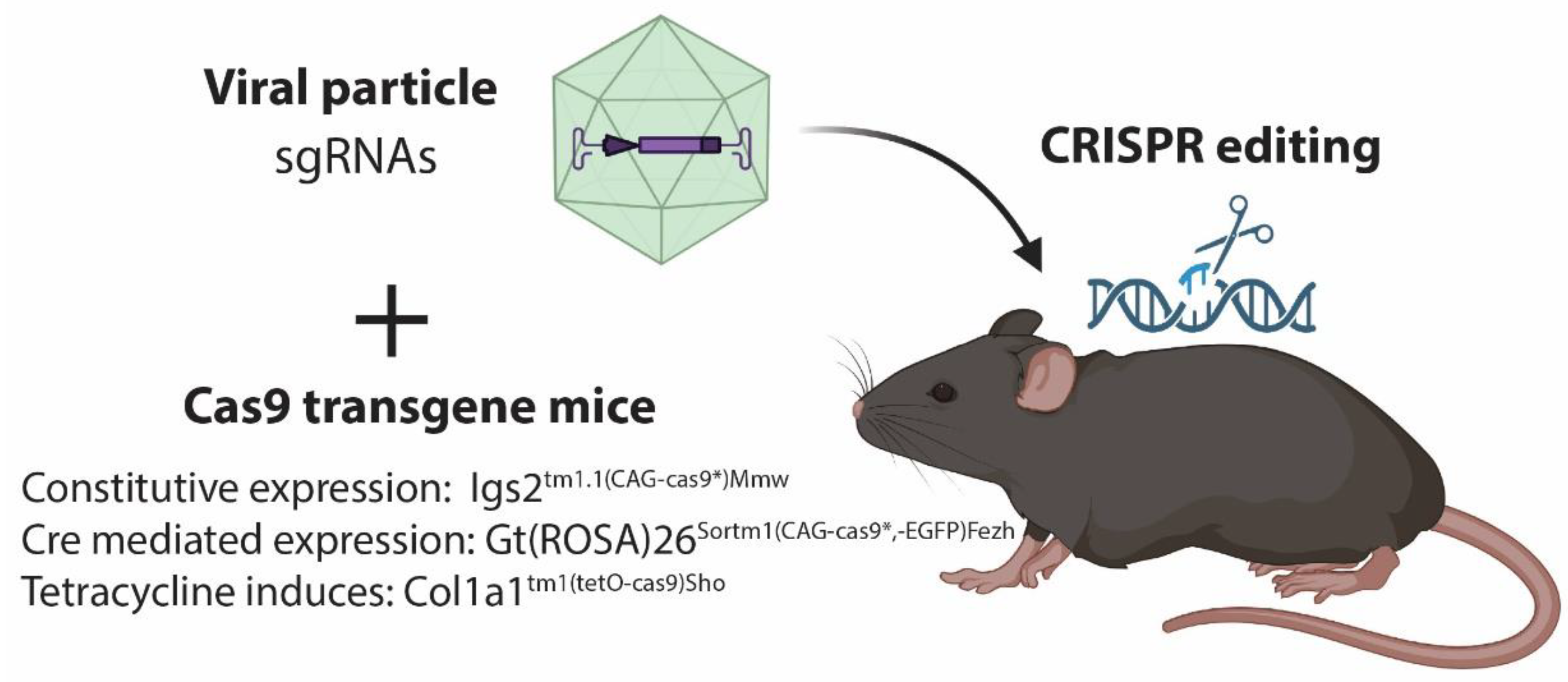
2.2. Viral Delivery of CRISPR/Cas9
2.3. Lipid Nanoparticle for In Vivo Delivery of CRISPR/Cas9
3. CRISPR Induced Tumors
3.1. Targeting Multiple Genes Simultaneously for Cancer Induction
3.2. Insertion and Deletion Analysis for Tumor Profiling
3.3. Chromosomal Rearrangement by CRISPR In Vivo
3.4. In Vivo CRISPR Screen
4. Pit Falls by CRISPR/Cas9 in In Vivo Cancer Modeling
4.1. Tumor Heterogeneity
4.2. Biased Indel Analysis and off Target Mutations
4.3. Delivery and Non-Intended Tumors Formation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Ravendran, S.; Mikkelsen, N.S.; Haldrup, J.; Cai, H.; Ding, X.; Paludan, S.R.; Thomsen, M.K.; Mikkelsen, J.G.; Bak, R.O. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 2022, 30, 2942–2951. [Google Scholar] [CrossRef]
- Hussain, S.-R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef]
- Thomsen, E.A.; Rovsing, A.B.; Anderson, M.V.; Due, H.; Huang, J.; Luo, Y.; Dybkaer, K.; Mikkelsen, J.G. Identification of BLNK and BTK as mediators of rituximab-induced programmed cell death by CRISPR screens in GCB-subtype diffuse large B-cell lymphoma. Mol. Oncol. 2020, 14, 1978–1997. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Murina, O.; Reijns, M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, K.; Doe, B.; Banerjee, R.; Yang, F.; Bradley, A. Chromosome engineering in zygotes with CRISPR / C as9. genesis 2016, 54, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, K.; Geuer, S.; Will, A.J.; Chan, W.L.; Paliou, C.; Borschiwer, M.; Harabula, I.; Wittler, L.; Franke, M.; Ibrahim, D.M.; et al. Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep. 2015, 10, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Öllinger, R.; Friedrich, M.; Ehmer, U.; Barenboim, M.; Steiger, K.; Heid, I.; Mueller, S.; Maresch, R.; Engleitner, T.; et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13982–13987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.; Cai, W.; Yang, G.R.; Bronson, R.T.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Chen, M.; Whitley, M.J.; Kuo, H.-C.; Xu, E.S.; Walens, A.; Mowery, Y.M.; van Mater, D.; Eward, W.C.; Cardona, D.M.; et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 2017, 8, 15999. [Google Scholar] [CrossRef] [PubMed]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7, 10770. [Google Scholar] [CrossRef] [Green Version]
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.W.; Tschida, B.R.; Moriarity, B.S.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelsen, M.; Riedel, M.; Cai, H.; Skaarup, S.; Alstrup, A.; Dagnæs-Hansen, F.; Luo, Y.; Jensen, U.; Hager, H.; Liu, Y.; et al. The CRISPR/Cas9 Minipig—A Transgenic Minipig to Produce Specific Mutations in Designated Tissues. Cancers 2021, 13, 3024. [Google Scholar] [CrossRef]
- Annunziato, S.; de Ruiter, J.R.; Henneman, L.; Brambillasca, C.S.; Lutz, C.; Vaillant, F.; Ferrante, F.; Drenth, A.P.; van der Burg, E.; Siteur, B.; et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat. Commun. 2019, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Counsell, J.R.; Asgarian, Z.; Meng, J.; Ferrer, V.; Vink, C.A.; Howe, S.J.; Waddington, S.N.; Thrasher, A.J.; Muntoni, F.; Morgan, J.E.; et al. Lentiviral vectors can be used for full-length dystrophin gene therapy. Sci. Rep. 2017, 7, 44775. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sánchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziato, S.; Kas, S.M.; Nethe, M.; Yücel, H.; Del Bravo, J.; Pritchard, C.; Bin Ali, R.; van Gerwen, B.; Siteur, B.; Drenth, A.P.; et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes Dev. 2016, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, S.-H.; Winters, I.P.; Wang, J.; Naranjo, S.; Dudgeon, C.; Tamburini, F.B.; Brady, J.J.; Yang, D.; Grüner, B.M.; Chuang, C.-H.; et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015, 29, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rivera, F.J.; Papagiannakopoulos, T.; Romero, R.; Tammela, T.; Bauer, M.R.; Bhutkar, A.; Joshi, N.; Subbaraj, L.; Bronson, R.T.; Xue, W.; et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 2014, 516, 428–431. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Kantor, B. Lentiviral Vectors for Delivery of Gene-Editing Systems Based on CRISPR/Cas: Current State and Perspectives. Viruses 2021, 13, 1288. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The First Effective In Vivo Gene Delivery Vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.; Peng, J.; Yan, Y.; Cao, P.; Wang, J.; Qiu, C.; Tang, L.; Liu, D.; Tang, L.; Jin, J.; et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014, 588, 3954–3958. [Google Scholar] [CrossRef] [Green Version]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowling, S.; Sritharan, D.; Osorio, F.G.; Nguyen, M.; Cheung, P.; Rodriguez-Fraticelli, A.; Patel, S.; Yuan, W.-C.; Fujiwara, Y.; Li, B.E.; et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 2020, 181, 1693–1694. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Bak, R.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Cheng, Q.; Min, Y.-L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef]
- Riedel, M.; Berthelsen, M.F.; Cai, H.; Haldrup, J.; Borre, M.; Paludan, S.R.; Hager, H.; Vendelbo, M.H.; Wagner, E.F.; Bakiri, L.; et al. In vivo CRISPR inactivation of Fos promotes prostate cancer progression by altering the associated AP-1 subunit Jun. Oncogene 2021, 40, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Berthelsen, M.; Leknes, S.; Riedel, M.; Pedersen, M.; Joseph, J.; Hager, H.; Vendelbo, M.; Thomsen, M. Comparative Analysis of Stk11/Lkb1 versus Pten Deficiency in Lung Adenocarcinoma Induced by CRISPR/Cas9. Cancers 2021, 13, 974. [Google Scholar] [CrossRef]
- Cai, H.; Agersnap, S.N.; Sjøgren, A.; Simonsen, M.K.; Blaavand, M.S.; Jensen, U.V.; Thomsen, M.K. In Vivo Application of CRISPR/Cas9 Revealed Implication of Foxa1 and Foxp1 in Prostate Cancer Proliferation and Epithelial Plasticity. Cancers 2022, 14, 4381. [Google Scholar] [CrossRef]
- Richardson, C.; Ray, G.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG Gene Fusion in Prostate Cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Blasco, R.B.; Karaca, E.; Ambrogio, C.; Cheong, T.-C.; Karayol, E.; Minero, V.G.; Voena, C.; Chiarle, R. Simple and Rapid In Vivo Generation of Chromosomal Rearrangements using CRISPR/Cas9 Technology. Cell Rep. 2014, 9, 1219–1227. [Google Scholar] [CrossRef]
- Cook, P.J.; Thomas, R.; Kannan, R.; de Leon, E.S.; Drilon, A.; Rosenblum, M.K.; Scaltriti, M.; Benezra, R.; Ventura, A. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat. Commun. 2017, 8, 15987. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Chow, R.D.; Ye, L.; Guzman, C.D.; Dai, X.; Dong, M.B.; Zhang, F.; Sharp, P.A.; Platt, R.J.; Chen, S. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR–mediated direct in vivo screening. Sci. Adv. 2018, 4, eaao5508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, R.D.; Guzman, C.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemberling, M.P.; Siklenka, K.; Rodriguez, E.; Tonn-Eisinger, K.R.; Barrera, A.; Liu, F.; Kantor, A.; Li, L.; Cigliola, V.; Hazlett, M.F.; et al. Transgenic mice for in vivo epigenome editing with CRISPR-based systems. Nat. Methods 2021, 18, 965–974. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, J.; Zhou, C.; Gao, N.; Rao, Z.; Li, H.; Hu, X.; Li, C.; Yao, X.; Shen, X.; et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR–dCas9-activator transgenic mice. Nat. Neurosci. 2018, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, K.J.; Wang, Y.J.; Dou, Z.; Wang, A.W.; Mosleh-Shirazi, E.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Berger, S.L.; Kaestner, K.H. Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology 2018, 68, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.-Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’keefe, D.D.; Núñez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507.e1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, C.J.; Adames, A.C.; Saur, D.; Rad, R. Tutorial: Design and execution of CRISPR in vivo screens. Nat. Protoc. 2022, 17, 1903–1925. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer evolution: Darwin and beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Gutkin, A.; Dammes, N.; Peer, D. Progress and challenges towards CRISPR/Cas clinical translation. Adv. Drug Deliv. Rev. 2020, 154, 176–186. [Google Scholar] [CrossRef]
- Riedel, M.; Berthelsen, M.F.; Bakiri, L.; Wagner, E.F.; Thomsen, M.K. Virus Delivery of CRISPR Guides to the Murine Prostate for Gene Alteration. J. Vis. Exp. 2018, 134, e57525. [Google Scholar] [CrossRef] [PubMed]
- Rieblinger, B.; Sid, H.; Duda, D.; Bozoglu, T.; Klinger, R.; Schlickenrieder, A.; Lengyel, K.; Flisikowski, K.; Flisikowska, T.; Simm, N.; et al. Cas9-expressing chickens and pigs as resources for genome editing in livestock. Proc. Natl. Acad. Sci. USA 2021, 118, e2022562118. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jin, Q.; Ruan, D.; Yang, Y.; Liu, Q.; Wu, H.; Zhou, Z.; Ouyang, Z.; Liu, Z.; Zhao, Y.; et al. Cre-dependent Cas9-expressing pigs enable efficient in vivo genome editing. Genome Res. 2017, 27, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Cai, H.; Stoltze, I.C.; Vendelbo, M.H.; Wagner, E.F.; Bakiri, L.; Thomsen, M.K. Targeting AP-1 transcription factors by CRISPR in the prostate. Oncotarget 2021, 12, 1956–1961. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomsen, M.K. Application of CRISPR for In Vivo Mouse Cancer Studies. Cancers 2022, 14, 5014. https://doi.org/10.3390/cancers14205014
Thomsen MK. Application of CRISPR for In Vivo Mouse Cancer Studies. Cancers. 2022; 14(20):5014. https://doi.org/10.3390/cancers14205014
Chicago/Turabian StyleThomsen, Martin K. 2022. "Application of CRISPR for In Vivo Mouse Cancer Studies" Cancers 14, no. 20: 5014. https://doi.org/10.3390/cancers14205014
APA StyleThomsen, M. K. (2022). Application of CRISPR for In Vivo Mouse Cancer Studies. Cancers, 14(20), 5014. https://doi.org/10.3390/cancers14205014