
The Vitamin D Receptor–BIM Axis Overcomes Cisplatin Resistance in Head and Neck Cancer

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Clinical Data Analysis
2.3. Cell Culture
2.4. Generation of Cisplatin-Resistant Cell Model
2.5. Cell Viability Assays
2.6. Fluorescence Microscopy
2.7. Cell Migration Assay/Wound-Healing Assays
2.8. Plasmids and Transfection
2.9. Protein Extraction, Immunoblot Analysis
2.10. Statistical Analysis
3. Results
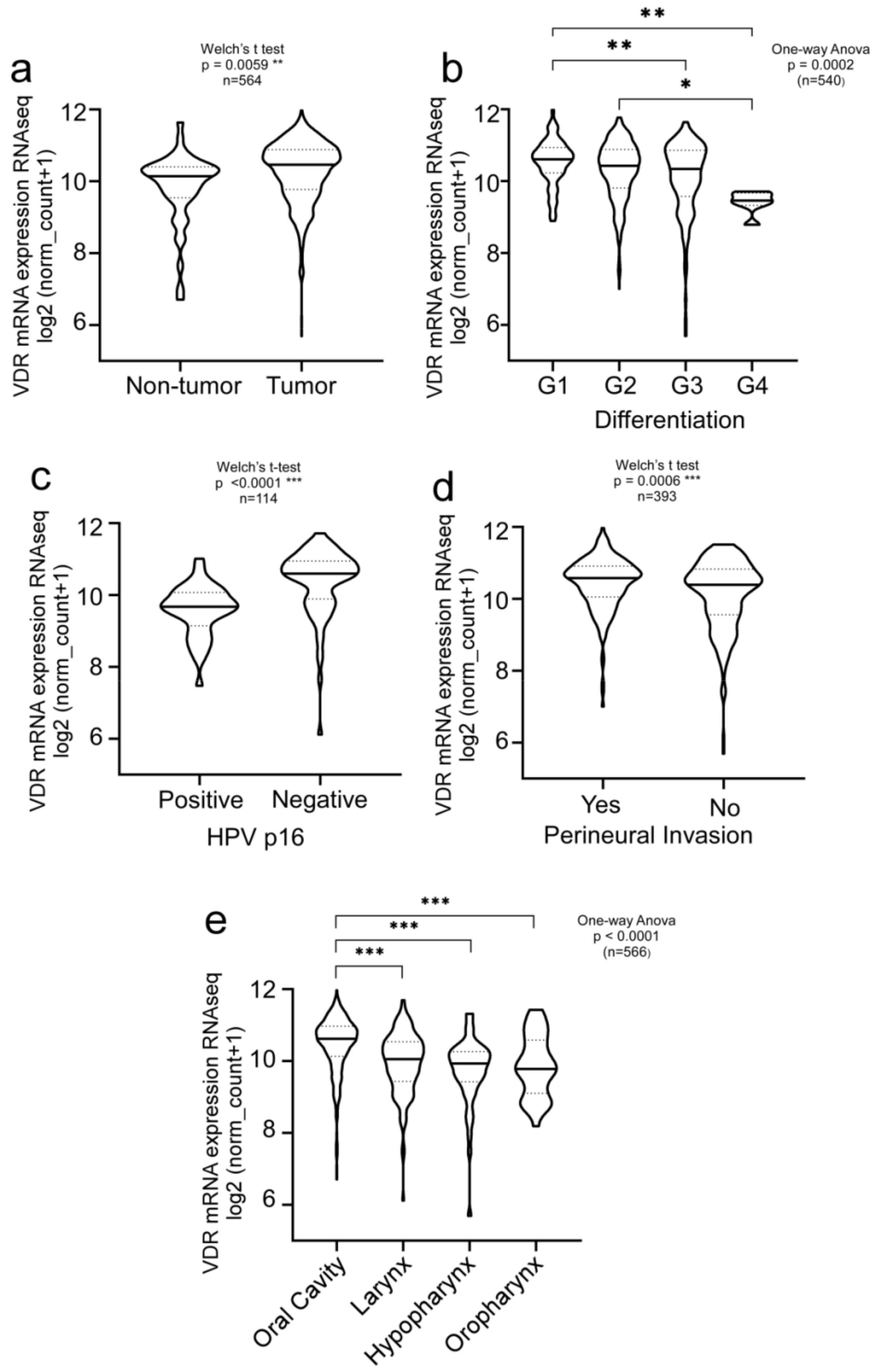
3.1. Clinical Relevance of Vitamin D Receptor (VDR) in HNSCC Patients
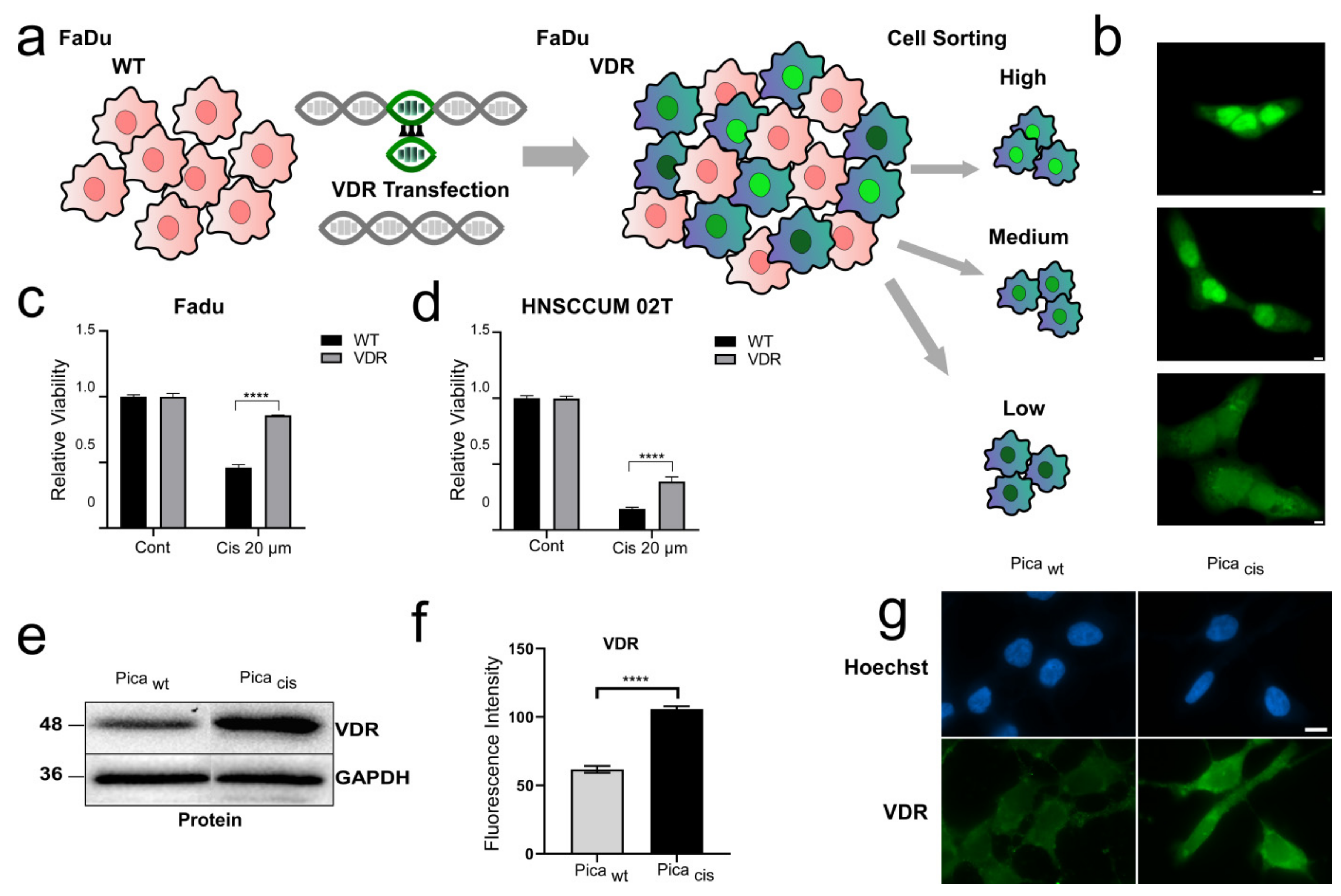
3.2. VDR Overexpression Contributes to Cisplatin Resistance
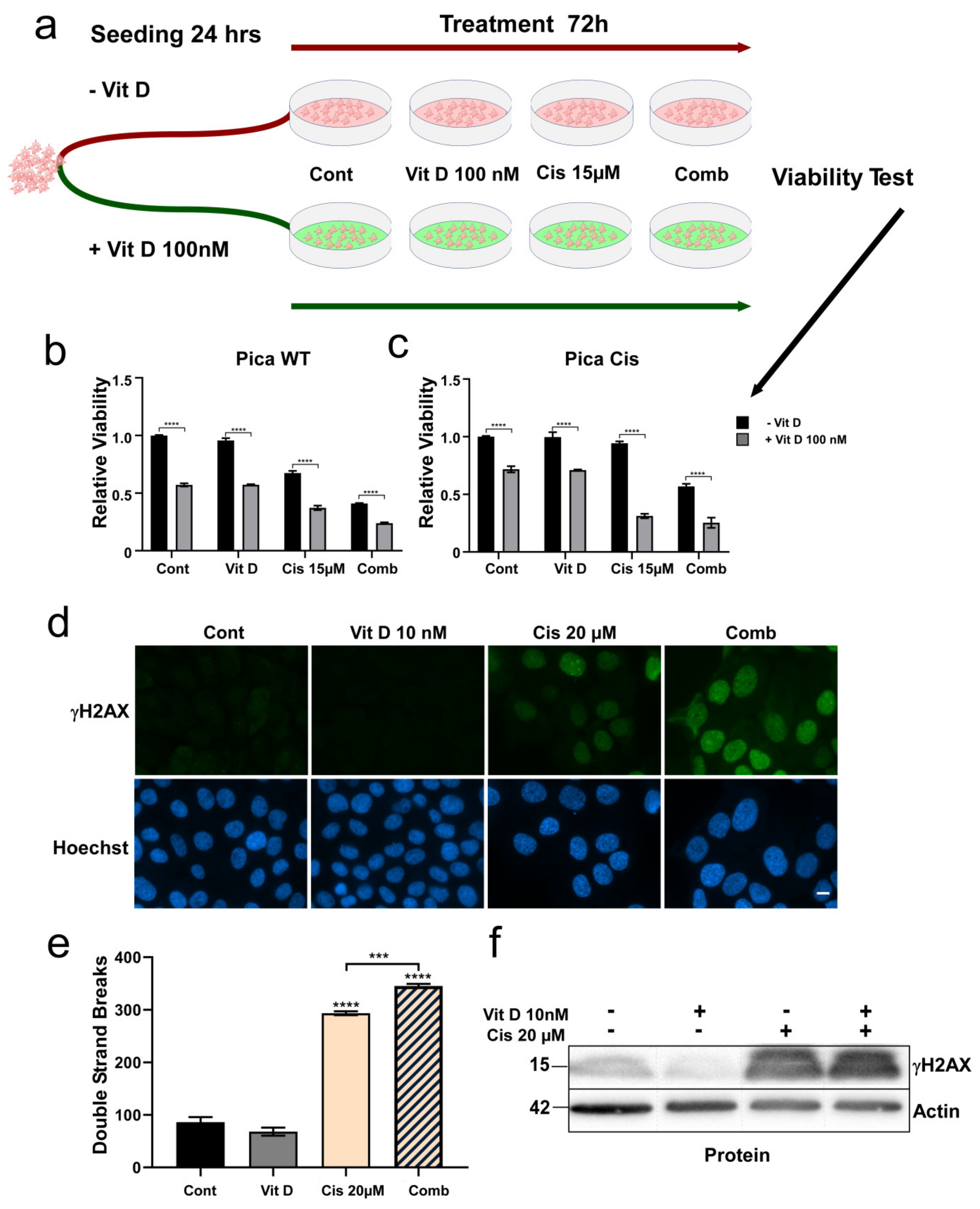
3.3. Ligand-Dependent VDR Expression/Activation Overcomes Cisplatin Resistance
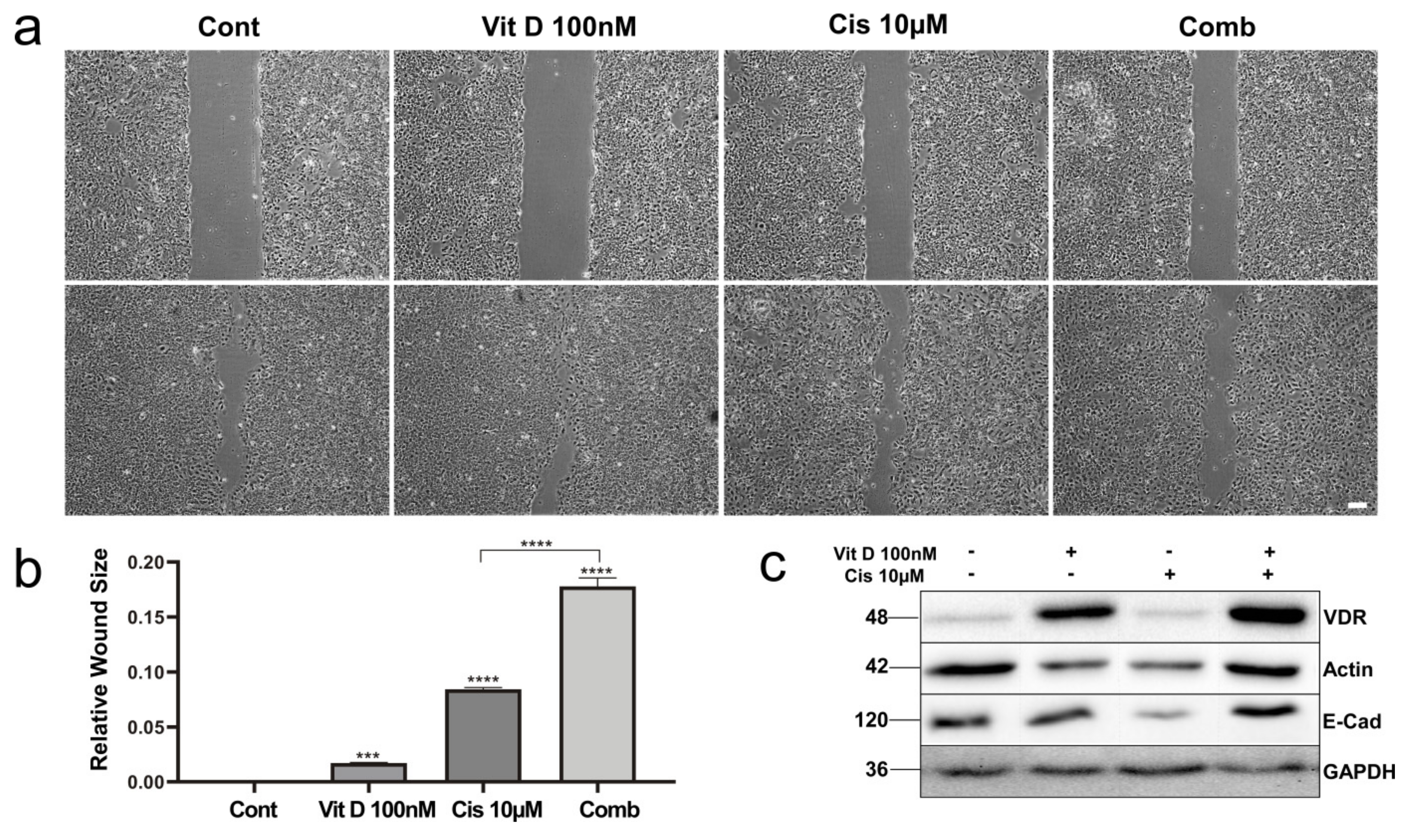
3.4. VitD Treatment Reduces Migration of HNSCC Cells
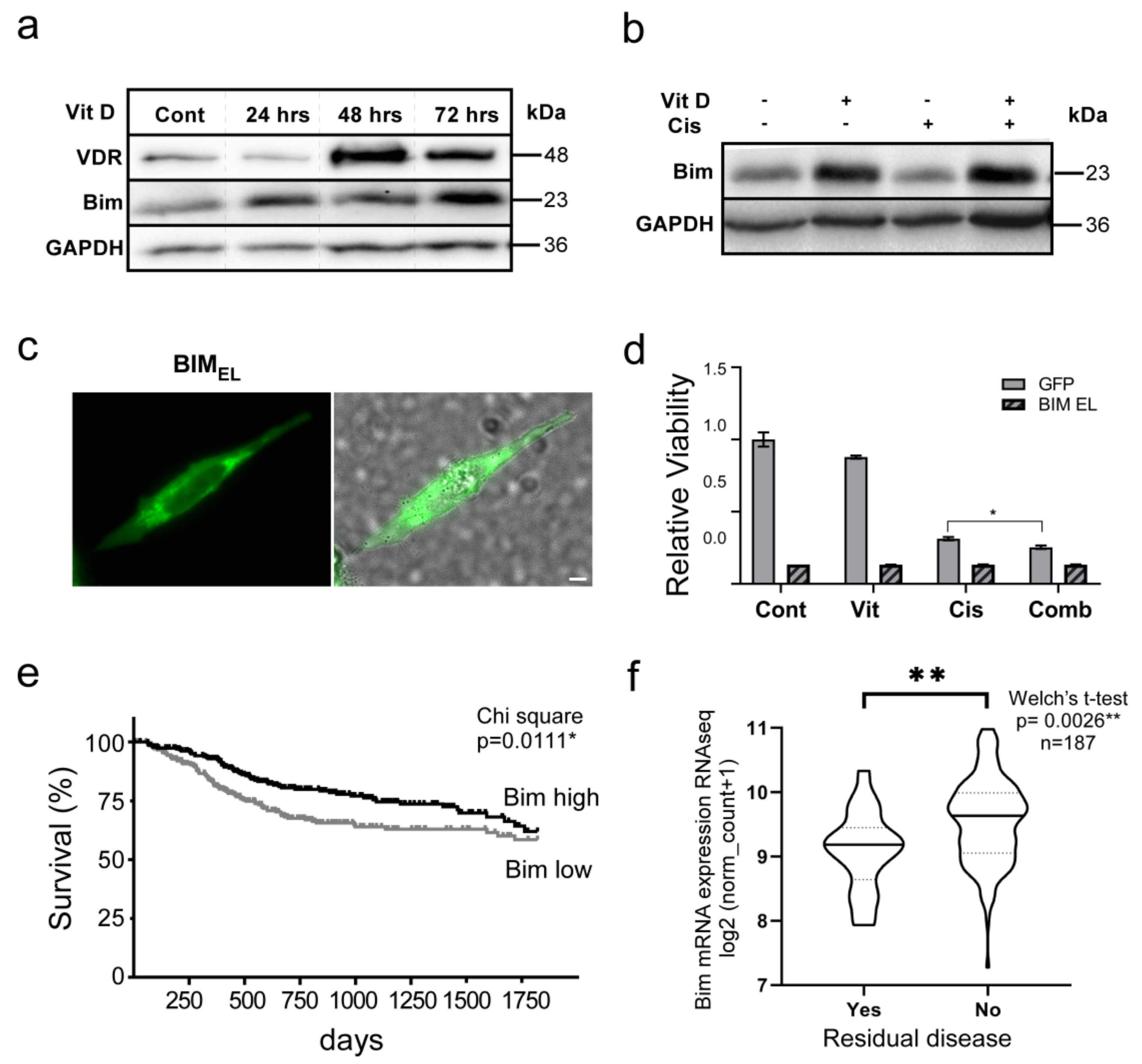
3.5. The VitD/VDR/BIM Axis Aids in Overcoming Cisplatin Resistance in Head and Neck Cancer
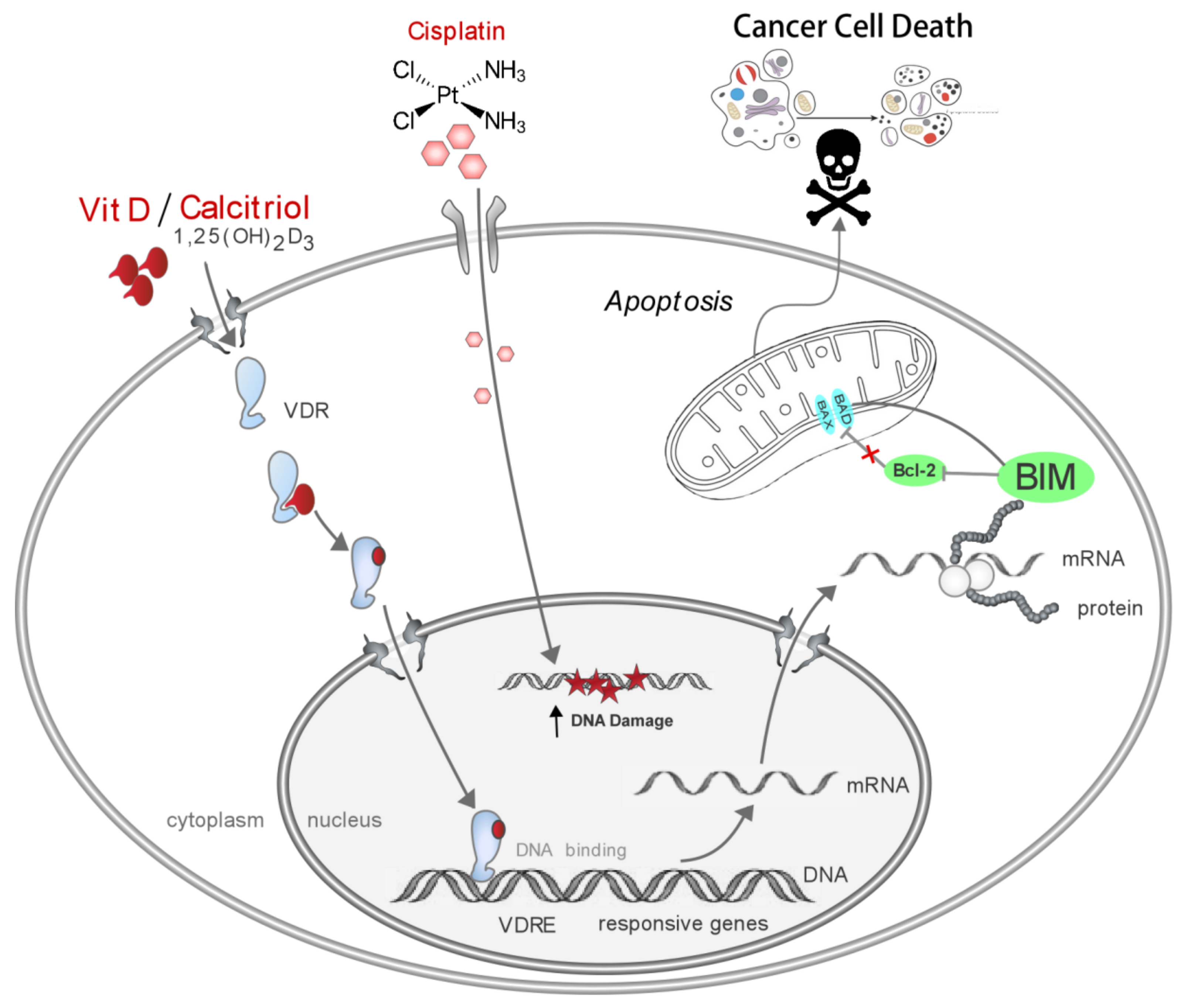
4. Discussion
5. Conclusions
6. Recommendations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siemer, S.; Fauth, T.; Scholz, P.; Al-Zamel, Y.; Khamis, A.; Gül, D.; Freudelsperger, L.; Wollenberg, B.; Becker, S.; Stauber, R.H.; et al. Profiling Cisplatin Resistance in Head and Neck Cancer: A Critical Role of the VRAC Ion Channel for Chemoresistance. Cancers 2021, 13, 4831. [Google Scholar] [CrossRef] [PubMed]
- Gul, D.; Schweitzer, A.; Khamis, A.; Knauer, S.K.; Ding, G.B.; Freudelsperger, L.; Karampinis, I.; Strieth, S.; Hagemann, J.; Stauber, R.H. Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer. Cancers 2022, 14, 2337. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ekanayake Weeramange, C.; Hughes, B.G.M.; Vasani, S.; Liu, Z.Y.; Warkiani, M.E.; Hartel, G.; Ladwa, R.; Thiery, J.P.; Kenny, L.; et al. Application of circulating tumour cells to predict response to treatment in head and neck cancer. Cell. Oncol. 2022, 45, 543–555. [Google Scholar] [CrossRef]
- Howren, M.B.; Christensen, A.J.; Pagedar, N.A. Problem alcohol and tobacco use in head and neck cancer patients at diagnosis: Associations with health-related quality of life. Support. Care Cancer 2022, 30, 8111–8118. [Google Scholar] [CrossRef]
- Forastiere, A.; Weber, R.; Ang, K. Treatment of head and neck cancer. N. Engl. J. Med. 2008, 358, 1076. [Google Scholar]
- Miserocchi, G.; Spadazzi, C.; Calpona, S.; De Rosa, F.; Usai, A.; De Vita, A.; Liverani, C.; Cocchi, C.; Vanni, S.; Calabrese, C.; et al. Precision Medicine in Head and Neck Cancers: Genomic and Preclinical Approaches. J. Pers. Med. 2022, 12, 854. [Google Scholar] [CrossRef]
- Siemer, S.; Bauer, T.A.; Scholz, P.; Breder, C.; Fenaroli, F.; Harms, G.; Dietrich, D.; Dietrich, J.; Rosenauer, C.; Barz, M.; et al. Targeting Cancer Chemotherapy Resistance by Precision Medicine-Driven Nanoparticle-Formulated Cisplatin. ACS Nano 2021, 15, 18541–18556. [Google Scholar] [CrossRef]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, A.W.; Meng, X.W.; Kaufmann, S.H. Anticancer therapy: Boosting the bang of Bim. J. Clin. Investig. 2008, 118, 3582–3584. [Google Scholar] [CrossRef] [Green Version]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and cytoplasmic survivin: Molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007, 67, 5999–6002. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, A.; Knauer, S.K.; Stauber, R.H. Nuclear receptors in head and neck cancer: Current knowledge and perspectives. Int. J. Cancer 2010, 126, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Font-Díaz, J.; Jiménez-Panizo, A.; Caelles, C.; Vivanco, M.d.; Pérez, P.; Aranda, A.; Estébanez-Perpiñá, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. Semin. Cancer Biol. 2021, 73, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Stauber, R.H.; Wunsch, D.; Knauer, S.K.; Fetz, V. An update on the pathobiological relevance of nuclear receptors for cancers of the head and neck. Histol. Histopathol. 2010, 25, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Chow, E.C.; Quach, H.P.; Groothuis, G.M.; Tirona, R.G.; Pang, K.S. Significance of the Vitamin D Receptor on Crosstalk with Nuclear Receptors and Regulation of Enzymes and Transporters. AAPS J. 2022, 24, 71. [Google Scholar] [CrossRef]
- Prufer, K.; Barsony, J. Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol. Endocrinol. 2002, 16, 1738–1751. [Google Scholar] [CrossRef]
- Gangwar, S.K.; Kumar, A.; Jose, S.; Alqahtani, M.S.; Abbas, M.; Sethi, G.; Kunnumakkara, A.B. Nuclear receptors in oral cancer-Emerging players in tumorigenesis. Cancer Lett. 2022, 536, 215666. [Google Scholar] [CrossRef]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear receptor crosstalk — defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef]
- Carlberg, C. Vitamin D and Its Target Genes. Nutrients 2022, 14, 1354. [Google Scholar] [CrossRef]
- Pludowski, P.; Takacs, I.; Boyanov, M.; Belaya, Z.; Diaconu, C.C.; Mokhort, T.; Zherdova, N.; Rasa, I.; Payer, J.; Pilz, S. Clinical Practice in the Prevention, Diagnosis and Treatment of Vitamin D Deficiency: A Central and Eastern European Expert Consensus Statement. Nutrients 2022, 14, 1483. [Google Scholar] [CrossRef]
- Wu, X.; Hu, W.; Lu, L.; Zhao, Y.; Zhou, Y.; Xiao, Z.; Zhang, L.; Zhang, H.; Li, X.; Li, W.; et al. Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm. Sin. B 2019, 9, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Janoušek, J.; Pilařová, V.; Macáková, K.; Nomura, A.; Veiga-Matos, J.; Silva, D.D.d.; Remião, F.; Saso, L.; Malá-Ládová, K.; Malý, J. Vitamin D: Sources, physiological role, biokinetics, deficiency, therapeutic use, toxicity, and overview of analytical methods for detection of vitamin D and its metabolites. Crit. Rev. Clin. Lab. Sci. 2022, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, S.; Di Nisio, A.; Magno, S.; Romano, F.; Barrea, L.; Colao, A.M.; Muscogiuri, G.; Savastano, S. Vitamin D deficiency: A potential risk factor for cancer in obesity? Int. J. Obes. 2022, 46, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Niedermaier, T.; Gredner, T.; Kuznia, S.; Schöttker, B.; Mons, U.; Brenner, H. Vitamin D supplementation to the older adult population in Germany has the cost-saving potential of preventing almost 30 000 cancer deaths per year. Mol. Oncol. 2021, 15, 1986–1994. [Google Scholar] [CrossRef]
- Vanhevel, J.; Verlinden, L.; Doms, S.; Wildiers, H.; Verstuyf, A. The role of vitamin D in breast cancer risk and progression. Endocr. -Relat. Cancer 2022, 29, R33–R55. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Q.; Liu, B.; Zhang, N.; Cheng, G. Vitamin D enhances radiosensitivity of colorectal cancer by reversing epithelial-mesenchymal transition. Front. Cell Dev. Biol. 2021, 9, 684855. [Google Scholar] [CrossRef]
- Pilz, S.; Trummer, C.; Theiler-Schwetz, V.; Grübler, M.R.; Verheyen, N.D.; Odler, B.; Karras, S.N.; Zittermann, A.; März, W. Critical appraisal of large vitamin D randomized controlled trials. Nutrients 2022, 14, 303. [Google Scholar] [CrossRef]
- Ibrahimovic, M.; Franzmann, E.; Mondul, A.M.; Weh, K.M.; Howard, C.; Hu, J.J.; Goodwin, W.J.; Kresty, L.A. Disparities in Head and Neck Cancer: A Case for Chemoprevention with Vitamin D. Nutrients 2020, 12, 2638. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Y.; Li, H.; Zhou, Y.; Zhang, Q.; Chen, R.; Jin, T.; Hu, K.; Li, S.; Wang, Y.; et al. Vitamin D promotes the cisplatin sensitivity of oral squamous cell carcinoma by inhibiting LCN2-modulated NF-κB pathway activation through RPS3. Cell Death Dis. 2019, 10, 936. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, W.K.; Flavin, R.; Kasperzyk, J.L.; Fiorentino, M.; Fang, F.; Lis, R.; Fiore, C.; Penney, K.L.; Ma, J.; Kantoff, P.W.; et al. Vitamin D receptor protein expression in tumor tissue and prostate cancer progression. J. Clin. Oncol. 2011, 29, 2378–2385. [Google Scholar] [CrossRef] [Green Version]
- Welkoborsky, H.J.; Jacob, R.; Riazimand, S.H.; Bernauer, H.S.; Mann, W.J. Molecular biologic characteristics of seven new cell lines of squamous cell carcinomas of the head and neck and comparison to fresh tumor tissue. Oncology 2003, 65, 60–71. [Google Scholar] [CrossRef]
- Rangan, S. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer 1972, 29, 117–121. [Google Scholar] [CrossRef]
- Habtemichael, N.; Heinrich, U.R.; Knauer, S.K.; Schmidtmann, I.; Bier, C.; Docter, D.; Brochhausen, C.; Helling, K.; Brieger, J.; Stauber, R.H.; et al. Expression analysis suggests a potential cytoprotective role of Birc5 in the inner ear. Mol. Cell. Neurosci. 2010, 45, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Bier, C.; Knauer, S.K.; Klapthor, A.; Schweitzer, A.; Rekik, A.; Kramer, O.H.; Marschalek, R.; Stauber, R.H. Cell-based Analysis of Structure-Function Activity of Threonine Aspartase 1. J. Biol. Chem. 2011, 286, 3007–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetz, V.; Knauer, S.K.; Bier, C.; Von Kries, J.P.; Stauber, R.H. Translocation biosensors–cellular system integrators to dissect CRM1-dependent nuclear export by chemicogenomics. Sensors 2009, 9, 5423–5445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Dong, M.; Sheng, W.; Liu, Q.; Yu, D.; Dong, Q.; Li, Q.; Wang, J. Expression of vitamin D receptor as a potential prognostic factor and therapeutic target in pancreatic cancer. Histopathology 2015, 67, 386–397. [Google Scholar] [CrossRef]
- Stauber, R.H.; Knauer, S.K.; Habtemichael, N.; Bier, C.; Unruhe, B.; Weisheit, S.; Spange, S.; Nonnenmacher, F.; Fetz, V.; Ginter, T.; et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget 2012, 3, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Docter, D.; Distler, U.; Storck, W.; Kuharev, J.; Wünsch, D.; Hahlbrock, A.; Knauer, S.K.; Tenzer, S.; Stauber, R.H. Quantitative profiling of the protein coronas that form around nanoparticles. Nat. Protoc. 2014, 9, 2030–2044. [Google Scholar] [CrossRef]
- Tolón, R.M.; Castillo, A.I.; Jiménez-Lara, A.M.; Aranda, A. Association with Ets-1 causes ligand-and AF2-independent activation of nuclear receptors. Mol. Cell. Biol. 2000, 20, 8793–8802. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, T.; Zheng, Y.; Fournier, P.G.J.; Murthy, S.; John, S.; Schillo, S.; Dunstan, C.R.; Mohammad, K.S.; Zhou, H.; Seibel, M.J.; et al. The vitamin D receptor is involved in the regulation of human breast cancer cell growth via a ligand-independent function in cytoplasm. Oncotarget 2017, 8, 26687–26701. [Google Scholar] [CrossRef] [PubMed]
- Kawa, S.; Yoshizawa, K.; Nikaido, T.; Kiyosawa, K. Inhibitory effect of 22-oxa-1,25-dihydroxyvitamin D3, maxacalcitol, on the proliferation of pancreatic cancer cell lines. J. Steroid Biochem. Mol. Biol. 2005, 97, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futawaka, K.; Tagami, T.; Fukuda, Y.; Koyama, R.; Nushida, A.; Nezu, S.; Yamamoto, H.; Imamoto, M.; Kasahara, M.; Moriyama, K. Transcriptional activation of the wild-type and mutant vitamin D receptors by vitamin D3 analogs. J. Mol. Endocrinol. 2016, 57, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.; Koiwa, F.; Ogata, H.; Shishido, K.; Sekiguchi, T.; Michihata, T.; Ogawa, H.; Mukai, M.; Takahashi, K.; Suzuki, R.; et al. Active vitamin D analogs, maxacalcitol and alfacalcidol, as maintenance therapy for mild secondary hyperparathyroidism in hemodialysis patients—A randomized study. Int. J. Clin. Pharmacol. Ther. 2014, 52, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Vuolo, L.; Di Somma, C.; Faggiano, A.; Colao, A. Vitamin D and cancer. Front. Endocrinol. 2012, 3, 58. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.S.; Oh, S.; Jang, B.; Yu, H.J.; Shin, J.A.; Cho, N.P.; Yang, I.H.; Won, D.H.; Kwon, H.J.; Hong, S.D.; et al. Silymarin and its active component silibinin act as novel therapeutic alternatives for salivary gland cancer by targeting the ERK1/2-Bim signaling cascade. Cell. Oncol. 2017, 40, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Masugi, Y.; Qian, Z.R.; Nishihara, R.; Liu, L.; Smith-Warner, S.A.; Keum, N.; Zhang, L.; Tchrakian, N.; Nowak, J.A.; et al. Tumor expression of calcium sensing receptor and colorectal cancer survival: Results from the nurses’ health study and health professionals follow-up study. Int. J. Cancer 2017, 141, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Chen, G.; King, A.N.; Jeon, C.K.; Christensen, P.J.; Zhao, L.; Simpson, R.U.; Thomas, D.G.; Giordano, T.J.; Brenner, D.E.; et al. Characterization of vitamin D receptor (VDR) in lung adenocarcinoma. Lung Cancer 2012, 77, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Kaldre, D.; Wang, T.T.; Fischer, J.; White, J.H.; Gleason, J.L. Optimization of histone deacetylase inhibitor activity in non-secosteroidal vitamin D-receptor agonist hybrids. Bioorg. Med. Chem. 2015, 23, 5035–5049. [Google Scholar] [CrossRef]
- Bunch, B.L.; Ma, Y.; Attwood, K.; Amable, L.; Luo, W.; Morrison, C.; Guru, K.A.; Woloszynska-Read, A.; Hershberger, P.A.; Trump, D.L.; et al. Vitamin D3 enhances the response to cisplatin in bladder cancer through VDR and TAp73 signaling crosstalk. Cancer Med. 2019, 8, 2449–2461. [Google Scholar] [CrossRef] [Green Version]
- Bochen, F.; Balensiefer, B.; Körner, S.; Bittenbring, J.T.; Neumann, F.; Koch, A.; Bumm, K.; Marx, A.; Wemmert, S.; Papaspyrou, G.; et al. Vitamin D deficiency in head and neck cancer patients–prevalence, prognostic value and impact on immune function. Oncoimmunology 2018, 7, e1476817. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Nachliely, M.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. Participation of vitamin D-upregulated protein 1 (TXNIP)-ASK1-JNK1 signalosome in the enhancement of AML cell death by a post-cytotoxic differentiation regimen. J. Steroid Biochem. Mol. Biol. 2019, 187, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Ligand-Independent Actions of the Vitamin D Receptor: More Questions Than Answers. JBMR Plus 2021, 5, e10578. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, A.; Jablonska, K.; Podhorska-Okolow, M.; Ugorski, M.; Dziegiel, P. Prolactin-induced protein (PIP)-characterization and role in breast cancer progression. Am. J. Cancer Res. 2018, 8, 2150. [Google Scholar]
- LaPensee, E.W.; Ben-Jonathan, N. Novel roles of prolactin and estrogens in breast cancer: Resistance to chemotherapy. Endocr. -Relat. Cancer 2010, 17, R91. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Luco, A.-L.; Camirand, A.; St-Arnaud, R.; Kremer, R. Vitamin D regulates CXCL12/CXCR4 and epithelial-to-mesenchymal transition in a model of breast cancer metastasis to lung. Endocrinology 2021, 162, bqab049. [Google Scholar] [CrossRef]
- Chandler, P.D.; Chen, W.Y.; Ajala, O.N.; Hazra, A.; Cook, N.; Bubes, V.; Lee, I.-M.; Giovannucci, E.L.; Willett, W.; Buring, J.E. Effect of vitamin D3 supplements on development of advanced cancer: A secondary analysis of the VITAL randomized clinical trial. JAMA Netw. Open 2020, 3, e2025850. [Google Scholar] [CrossRef]
- Suares, A.; Tapia, C.; González-Pardo, V. Antineoplastic effect of 1α,25(OH)(2)D(3) in spheroids from endothelial cells transformed by Kaposi’s sarcoma-associated herpesvirus G protein coupled receptor. J. Steroid Biochem. Mol. Biol. 2019, 186, 122–129. [Google Scholar] [CrossRef]
- Lisse, T.S.; Hewison, M. Vitamin D: A new player in the world of mTOR signaling. Cell Cycle 2011, 10, 1888–1889. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamis, A.; Gül, D.; Wandrey, M.; Lu, Q.; Knauer, S.K.; Reinhardt, C.; Strieth, S.; Hagemann, J.; Stauber, R.H. The Vitamin D Receptor–BIM Axis Overcomes Cisplatin Resistance in Head and Neck Cancer. Cancers 2022, 14, 5131. https://doi.org/10.3390/cancers14205131
Khamis A, Gül D, Wandrey M, Lu Q, Knauer SK, Reinhardt C, Strieth S, Hagemann J, Stauber RH. The Vitamin D Receptor–BIM Axis Overcomes Cisplatin Resistance in Head and Neck Cancer. Cancers. 2022; 14(20):5131. https://doi.org/10.3390/cancers14205131
Chicago/Turabian StyleKhamis, Aya, Désirée Gül, Madita Wandrey, Qiang Lu, Shirley K. Knauer, Christoph Reinhardt, Sebastian Strieth, Jan Hagemann, and Roland H. Stauber. 2022. "The Vitamin D Receptor–BIM Axis Overcomes Cisplatin Resistance in Head and Neck Cancer" Cancers 14, no. 20: 5131. https://doi.org/10.3390/cancers14205131
APA StyleKhamis, A., Gül, D., Wandrey, M., Lu, Q., Knauer, S. K., Reinhardt, C., Strieth, S., Hagemann, J., & Stauber, R. H. (2022). The Vitamin D Receptor–BIM Axis Overcomes Cisplatin Resistance in Head and Neck Cancer. Cancers, 14(20), 5131. https://doi.org/10.3390/cancers14205131