
Geostatistical Modeling and Heterogeneity Analysis of Tumor Molecular Landscape




Abstract
:Simple Summary
Abstract
1. Introduction
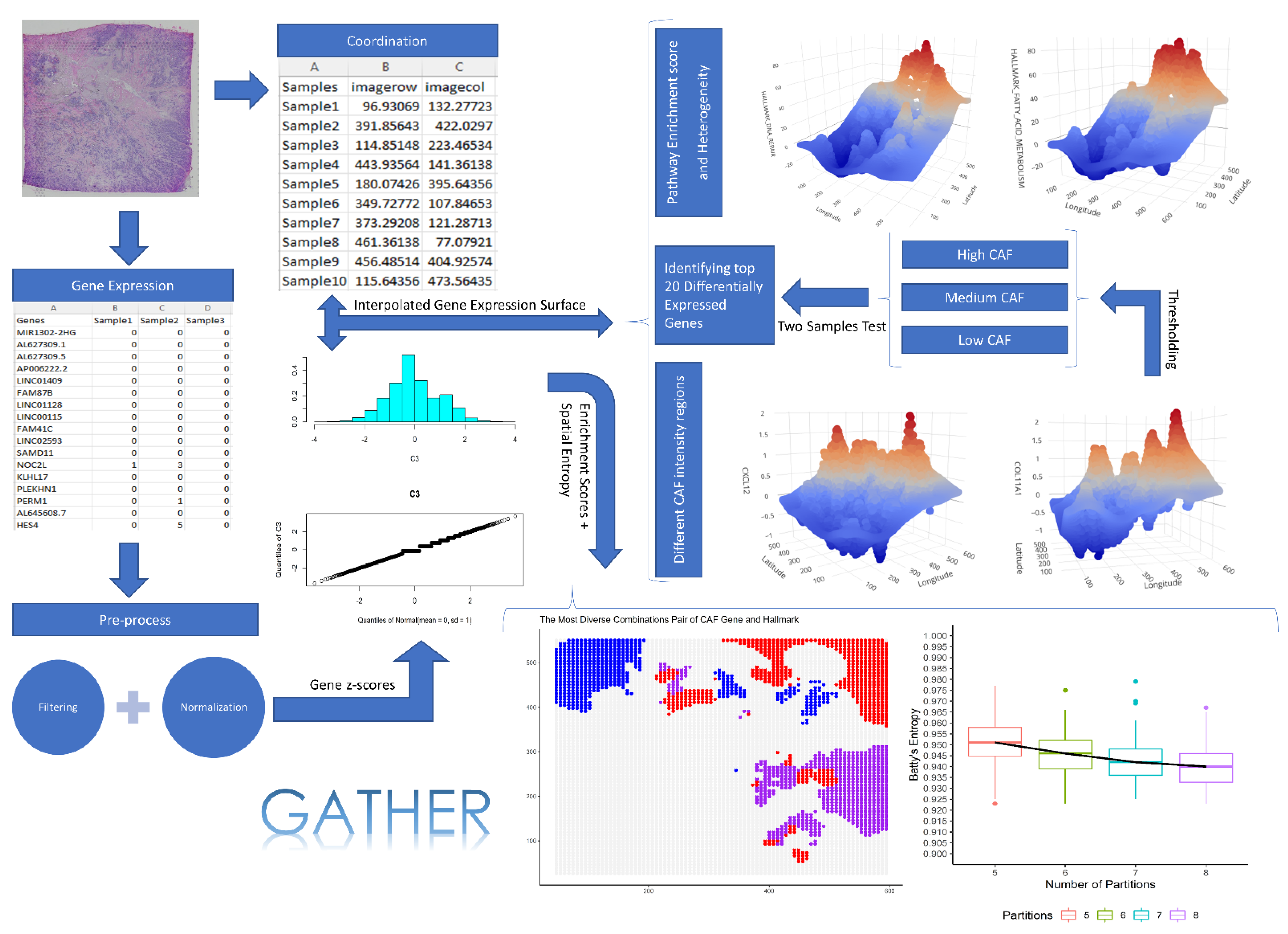
2. Data and Methods
2.1. Data
2.2. Constructing Gene Expression Landscape by Kriging
2.3. Test of Spatial Differential Expression of Genes
2.4. Spatial Analysis of Hallmark Gene Sets of Cancer
2.4.1. Gene set Enrichment Landscape Construction
2.4.2. Cancer Hallmark Gene Set Enrichment
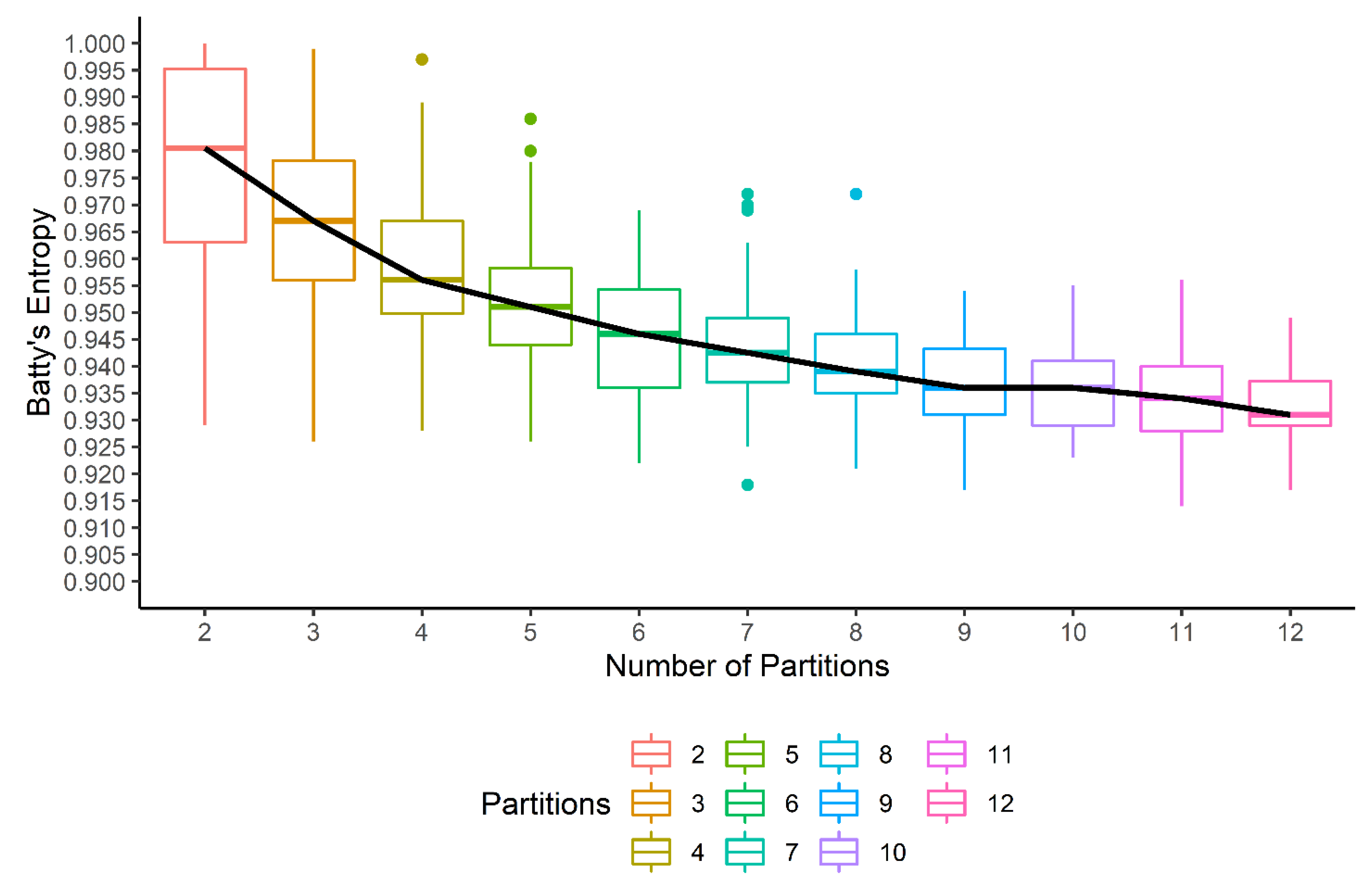
2.5. Spatial Entropy of a Tumor Sample
2.5.1. Calculating Phenotypic Diversity
2.5.2. Heterogeneity of Gene Set Enrichment in a Phenotypic Context
2.6. Software
3. Results
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R. Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Liu, L.; Almendro, V.; Kuang, Y.; Paweletz, C.; Sakr, R.A.; Weigelt, B.; Hanker, A.B.; Chandarlapaty, S.; King, T.A. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015, 47, 1212–1219. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, G.; Pujadas, E.; Goutsias, J.; Feinberg, A.P. Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat. Genet. 2017, 49, 719–729. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Gittelman, R.; Gao, J.; Zhang, J.; Yusko, E.C.; Wu, C.-J.; Emerson, R.; Zhang, J.; Tipton, C.; Li, J. TCR Repertoire Intratumor Heterogeneity in Localized Lung Adenocarcinomas: An Association with Predicted Neoantigen Heterogeneity and Postsurgical RecurrenceTCR Intratumor Heterogeneity and Relapse in Lung Cancer. Cancer Discov. 2017, 7, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Cortez, E.; Roswall, P.; Pietras, K. Functional subsets of mesenchymal cell types in the tumor microenvironment. Semin. Cancer Biol. 2014, 25, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Anderberg, C.; Pietras, K. On the Origin of Cancer-Associated Fibroblasts; Taylor & Francis: Oxfordshire, UK, 2009. [Google Scholar]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Che, G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett. 2017, 13, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.H.; Hwang-Verslues, W.W.; Chang, Y.C.; Chen, C.C.; Hsiao, M.; Jeng, Y.M.; Chang, K.J.; Lee, E.Y.; Shew, J.Y.; Lee, W.H. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/β-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.-C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710. [Google Scholar] [CrossRef] [Green Version]
- Cuiffo, B.G.; Karnoub, A.E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adhes. Migr. 2012, 6, 220–230. [Google Scholar] [CrossRef]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Rye, I.H.; Trinh, A.; Sætersdal, A.B.; Nebdal, D.; Lingjærde, O.C.; Almendro, V.; Polyak, K.; Børresen-Dale, A.L.; Helland, Å.; Markowetz, F. Intratumor heterogeneity defines treatment-resistant HER 2+ breast tumors. Mol. Oncol. 2018, 12, 1838–1855. [Google Scholar] [CrossRef] [Green Version]
- Kalisky, T.; Oriel, S.; Bar-Lev, T.H.; Ben-Haim, N.; Trink, A.; Wineberg, Y.; Kanter, I.; Gilad, S.; Pyne, S. A brief review of single-cell transcriptomic technologies. Brief. Funct. Genom. 2018, 17, 64–76. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Rong, D.; Zhang, H.; Shi, X.; Yang, W.; Zheng, W.; Sun, G.; Wu, F.; Cao, H. Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther.-Oncolytics 2021, 21, 183–206. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem. Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X. Single-cell RNA sequencing reveals a dynamic stromal niche that supports tumor growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical OutcomesStromal Fibroblasts and Therapy Resistance. Cancer Res. 2016, 76, 6495–6506. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.-H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.-C. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Gillies, R.J.; Brown, J.S.; Anderson, A.R.; Gatenby, R.A. Eco-evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nat. Rev. Cancer 2018, 18, 576–585. [Google Scholar] [CrossRef]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection ForcesDarwinian Dynamics of Intratumoral Heterogeneity. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goovaerts, P. Geostatistical approaches for incorporating elevation into the spatial interpolation of rainfall. J. Hydrol. 2000, 228, 113–129. [Google Scholar] [CrossRef]
- Altieri, L.; Cocchi, D.; Roli, G. Advances in spatial entropy measures. Stoch. Environ. Res. Risk Assess. 2019, 33, 1223–1240. [Google Scholar] [CrossRef]
- Ramdas, A.; Trillos, N.G.; Cuturi, M. On wasserstein two-sample testing and related families of nonparametric tests. Entropy 2017, 19, 47. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Altieri, L.; Cocchi, D.; Roli, G. Spatial entropy for biodiversity and environmental data: The R-package SpatEntropy. Environ. Model. Softw. 2021, 144, 105149. [Google Scholar] [CrossRef]
- Batty, M. Entropy in spatial aggregation. Geogr. Anal. 1976, 8, 1–21. [Google Scholar] [CrossRef]
- MacArthur, R. Fluctuations of animal populations and a measure of community stability. Ecology 1955, 36, 533–536. [Google Scholar] [CrossRef]
- Leibovici, D.G.; Birkin, M.H. On geocomputational determinants of entropic variations for urban dynamics studies. Geogr. Anal. 2015, 47, 193–218. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Frieda, K.L.; Linton, J.M.; Hormoz, S.; Choi, J.; Chow, K.-H.K.; Singer, Z.S.; Budde, M.W.; Elowitz, M.B.; Cai, L. Synthetic recording and in situ readout of lineage information in single cells. Nature 2017, 541, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Findlay, G.M.; Gagnon, J.A.; Horwitz, M.S.; Schier, A.F.; Shendure, J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 2016, 353, aaf7907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Lubeck, E.; Zhou, W.; Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 2016, 92, 342–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.; Li, C.; Amamoto, R. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442–450. [Google Scholar] [CrossRef]
- Alemany, A.; Florescu, M.; Baron, C.S.; Peterson-Maduro, J.; Van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 2018, 556, 108–112. [Google Scholar] [CrossRef]
- Spanjaard, B.; Hu, B.; Mitic, N.; Olivares-Chauvet, P.; Janjuha, S.; Ninov, N.; Junker, J.P. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol. 2018, 36, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Codeluppi, S.; Borm, L.E.; Zeisel, A.; La Manno, G.; van Lunteren, J.A.; Svensson, C.I.; Linnarsson, S. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 2018, 15, 932–935. [Google Scholar] [CrossRef]
- Peterson, R.A.; Peterson, M.R.A. Package ‘bestNormalize’, Normalizing Transformation Functions. R Package Version; The Comprehensive R Archive Network (CRAN) Repository. 2022, Volume 1. Available online: https://petersonr.github.io/bestNormalize/ (accessed on 10 September 2022).
- Haining, R.P.; Haining, R. Spatial Data Analysis: Theory and Practice; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Altieri, L.; Cocchi, D.; Roli, G. SpatEntropy: Spatial Entropy Measures in R. arXiv 2018, arXiv:1804.05521. [Google Scholar]
- Leibovici, D.G. Defining spatial entropy from multivariate distributions of co-occurrences. In Proceedings of the International Conference on Spatial Information Theory, Landéda, France, 21–25 September 2009; pp. 392–404. [Google Scholar]
- Gribov, A.; Sill, M.; Lück, S.; Rücker, F.; Döhner, K.; Bullinger, L.; Benner, A.; Unwin, A. SEURAT: Visual analytics for the integrated analysis of microarray data. BMC Med. Genom. 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Using the bestNormalize Package. Available online: https://cran.r-project.org/web/packages/bestNormalize/vignettes/bestNormalize.html (accessed on 10 September 2022).
- Hiemstra, P.; Hiemstra, M.P. Package ‘automap’. Compare 2013, 105, 10. [Google Scholar]
- Schefzik, R.; Flesch, J.; Goncalves, A. Fast identification of differential distributions in single-cell RNA-sequencing data with waddR. Bioinformatics 2021, 37, 3204–3211. [Google Scholar] [CrossRef]
- Plot3D: Tools for Plotting 3-D and 2-D Data. Available online: https://cran.microsoft.com/snapshot/2016-03-28/web/packages/plot3D/vignettes/plot3D.pdf (accessed on 10 September 2022).
- Sievert, C. Interactive Web-Based Data Visualization With R, Plotly, and Shiny; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Robinson, M.; McCarthy, D.; Chen, Y.; Smyth, G.K. edgeR: Differential expression analysis of digital gene expression data. User’s Guide 2013, 26, 139–140. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Aliwaini, S.; Lubbad, A.M.; Shourfa, A.; Hamada, H.A.; Ayesh, B.; Abu Tayem, H.E.M.; Abu Mustafa, A.; Abu Rouk, F.; Redwan, M.M.; Al-Najjar, M. Overexpression of TBX3 transcription factor as a potential diagnostic marker for breast cancer. Mol. Clin. Oncol. 2019, 10, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Willmer, T.; Cooper, A.; Peres, J.; Omar, R.; Prince, S. The T-Box transcription factor 3 in development and cancer. Biosci. Trends 2017, 11, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Villa, F.; García-Ocaña, M.; Galván, J.A.; García-Martínez, J.; García-Pravia, C.; Menéndez-Rodríguez, P.; Rey, C.G.-d.; Barneo-Serra, L.; de Los Toyos, J.R. COL11A1/(pro) collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumor Biol. 2015, 36, 2213–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.T.; Tan, Y.J.; Falasca, M.; Oon, C.E. Cancer-associated fibroblasts: Epigenetic regulation and therapeutic intervention in breast cancer. Cancers 2020, 12, 2949. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Wang, L.; Shi, L.; Yun, F.; Liu, X.; Chen, Y.; Chen, C.; Ren, Y.; Jia, Y. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 2019, 24, 38. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal adhesion kinase fine tunes multifaced signals toward breast cancer progression. Cancers 2021, 13, 645. [Google Scholar] [CrossRef]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef]
- Luo, M.; Guan, J.-L. Focal adhesion kinase: A prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010, 289, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.M.; Rodriguez, Y.A.; Jeong, K.; Ahn, E.-Y.E.; Lim, S.-T.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Cheng, Y.-K.; Randles, A.; Itzkovitz, S.; Marusyk, A.; Ametller, E.; Gonzalez-Farre, X.; Muñoz, M.; Russnes, H.G.; Helland, Å. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014, 6, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Riaz, N.; Desrichard, A.; Şenbabaoğlu, Y.; Hakimi, A.A.; Makarov, V.; Reis-Filho, J.S.; Chan, T.A. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016, 7, 10051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Korenchan, D.E.; Flavell, R.R. Spatiotemporal pH heterogeneity as a promoter of cancer progression and therapeutic resistance. Cancers 2019, 11, 1026. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A. Evolution of metastases in space and time under immune selection. Cell 2018, 175, 751–765.e16. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-R.; Fallahi-Sichani, M.; Sorger, P.K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015, 6, 8390. [Google Scholar] [CrossRef] [Green Version]
- Gotway, C.A.; Wolfinger, R.D. Spatial prediction of counts and rates. Stat. Med. 2003, 22, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Khodayari Moez, E.; Hajihosseini, M.; Andrews, J.L.; Dinu, I. Longitudinal linear combination test for gene set analysis. BMC Bioinform. 2019, 20, 650. [Google Scholar] [CrossRef] [PubMed]
- Vatanpour, S.; Pyne, S.; Leite, A.P.; Dinu, I. Gene set analysis and reduction for a continuous phenotype: Identifying markers of birth weight variation based on embryonic stem cells and immunologic signatures. Comput. Biol. Med. 2019, 113, 103389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pyne, S.; Dinu, I. Gene set enrichment analysis for multiple continuous phenotypes. BMC Bioinform. 2014, 15, 260. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
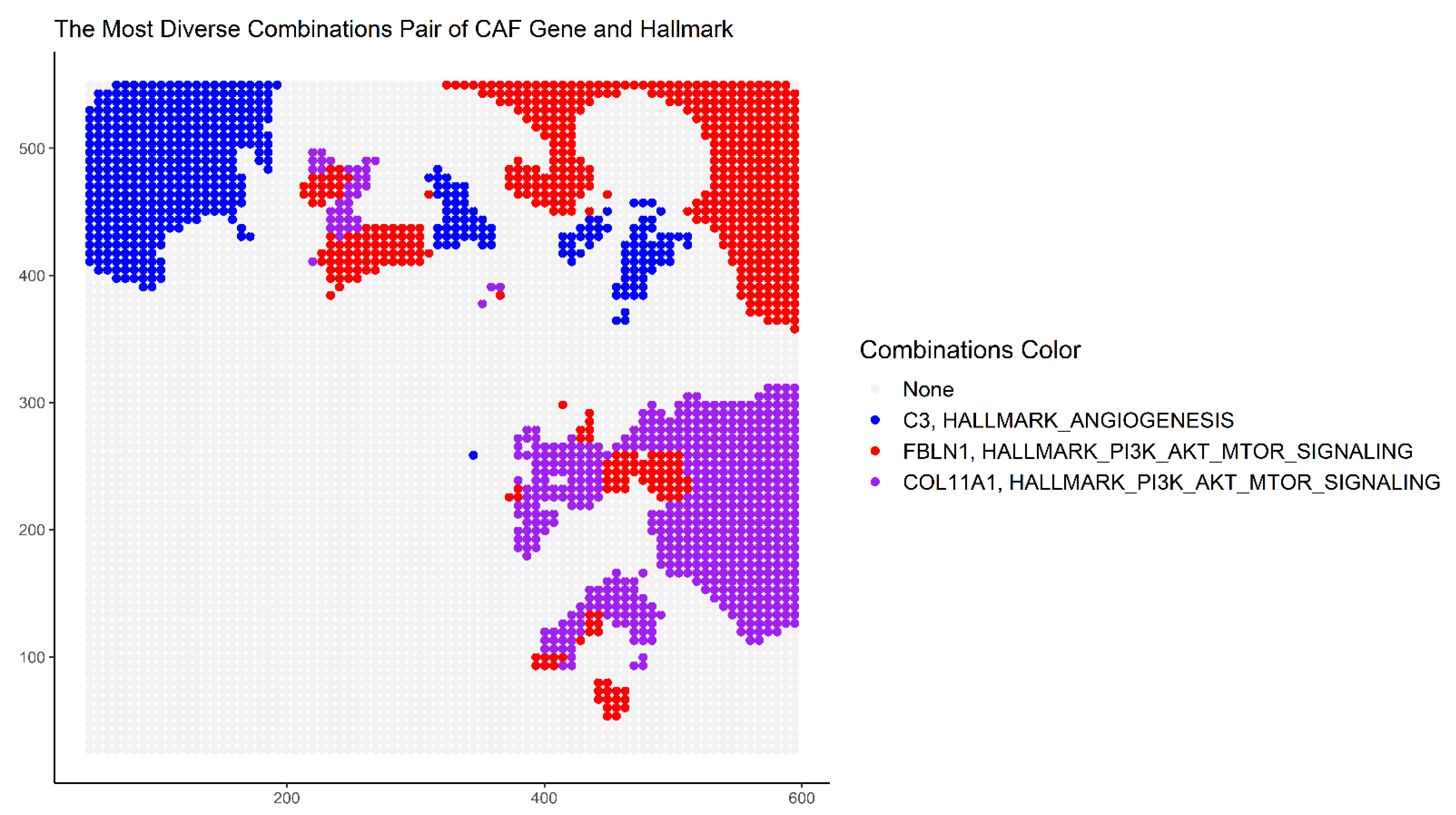{kind=link}
| CAF Marker | High CAF Z ≥ 1 N | Medium CAF 0.5 < Z < 1 N | Low CAF Z ≤ 0.5 N | The Top 20 Most Common Expressed Genes in 100-Times Permutation at q < 0.2 (N = 50 Random Samples for All Groups) | Median of Batty’s Spatial Entropy | |
|---|---|---|---|---|---|---|
| High CAF vs. Medium CAF | Medium CAF vs. Low CAF | |||||
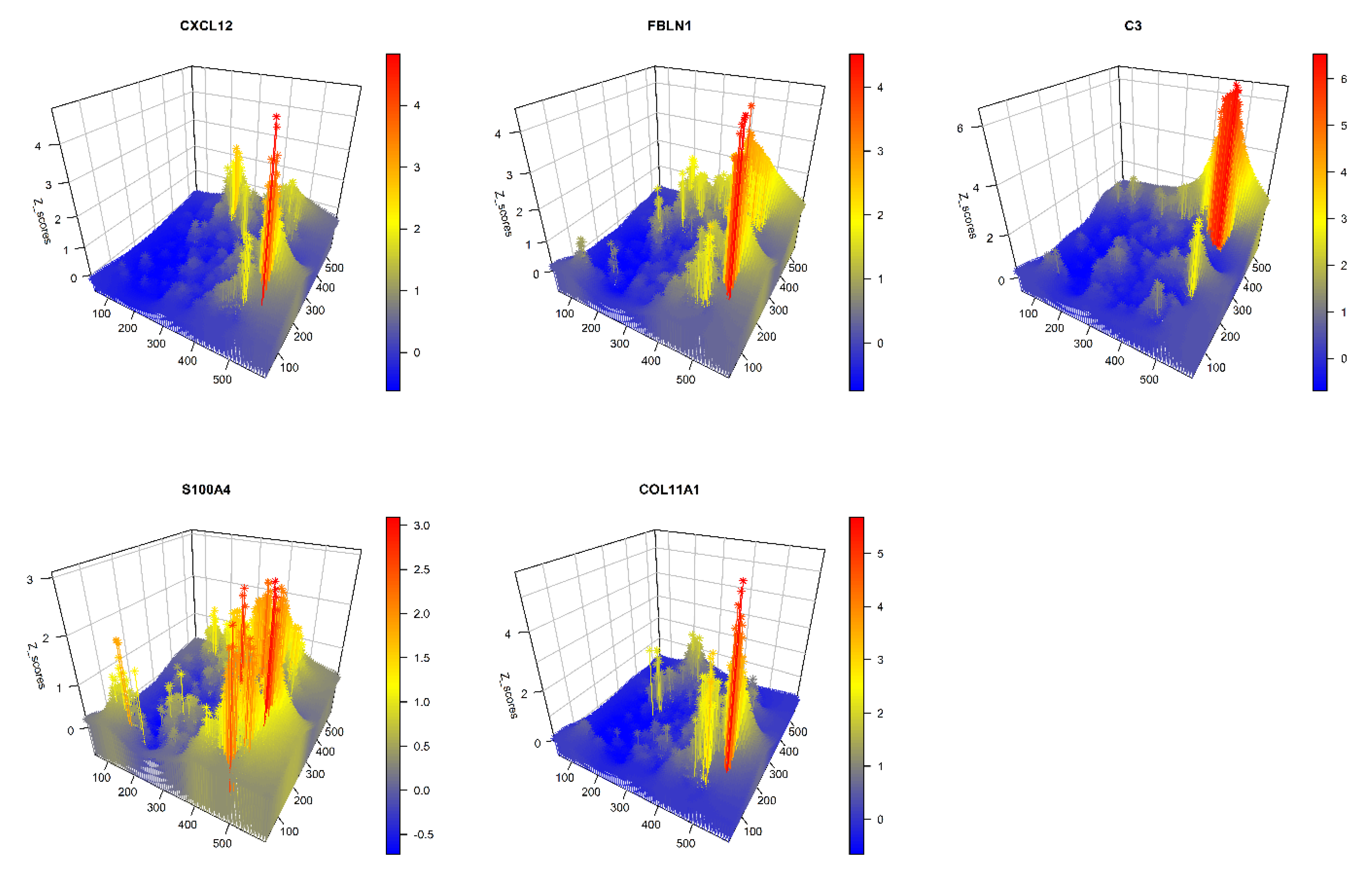
| COL11A1 | 190 | 3600 | 535 | MMP11, COL1A2, FN1, DCN, S100A6, CTSK, COL3A1, COL1A1, TIMP3, LUM, SDC1, B2M, S100A4, COL10A1, LGALS1, COL5A2, SERPINF1, SPARC, HLA.A, CTSD | COL1A2, ASPN, DCN, SDC1, LGALS1, COL1A1, SPARC, TAGLN, HTRA3, POSTN, COL5A1, PRSS23, AEBP1, CALD1, ACTA2, COL5A2, PTMS, FN1, COL6A2, FSTL1 | 0.983 |
| S100A4 | 223 | 3600 | 502 | LGALS1, S100A6, COL3A1, ACTB, HTRA1, S100A10, TAGLN, COL6A3, CD74, CRABP2, POSTN, TMSB10, HLA.DRB1, PALLD, CLU, SPARC, COL1A1, PTMS, COL6A1, SDC1 | FSTL1, SERPING1, COL3A1, COL6A2, FTL, ISLR, LGALS1, S100A6, SPARC, TAGLN, C1S, CILP, COL1A1, COL6A1, DCN, FLNA, HLA.DPA1, HLA.DPB1, PCOLCE, PTMS | 0.982 |
| CXCL12 | 141 | 3553 | 631 | COL6A2, DCN, MMP2, HSPG2, NBL1, SERPING1, SERPINF1, COL6A1, ISLR, AEBP1, ASPN, SPARC, LUM, COL5A2, THY1, LRP1, COL1A1, MMP11, COL3A1, RARRES2 | ACTB, ASPN, BGN, CALD1, CILP, COL1A1, COL3A1, COL5A1, COL6A2, DCN, FLNA, FN1, FSTL1, HTRA3, LGALS1, LUM, S100A6, SDC1, SPARC, TAGLN | 0.983 |
| C3 | 206 | 3501 | 618 | HLA.DRA, FTL, CYBA, HLA.DPB1, APOE, HLA.DPA1, CD74, A2M, RPL13, IFI27, LAPTM5, TYROBP, CTSB, VIM, ACTB, HLA.E, SERPING1, HLA.DRB1, PSAP, TMSB10 | APOE, COL5A1, FSTL1, SPARC, BGN, COL5A2, GPRC5A, PRCP, AP2M1, EDF1, HLA.DPA1, PITX1, ARHGAP1, COL6A1, COL6A2, CYB561, ATP5IF1, CD81, COL1A1, COL1A2 | 0.983 |
| FBLN1 | 288 | 3449 | 588 | LUM, COL3A1, COL6A2, FTL, C3, IFI27, COL1A1, COL1A2, MMP2, SERPING1, COL6A1, LRP1, SERPINF1, COL6A3, LGALS1, SPARC, FN1, ACTB, HTRA1, IFITM3 | COL3A1, DCN, SPARC, CILP, COL5A1, FN1, LGALS1, MYL9, ACTB, ASPN, CALD1, COL1A1, COL6A2, MMP11, POSTN, S100A6, TAGLN, TPM4, COL1A2, COL6A1 | 0.982 |
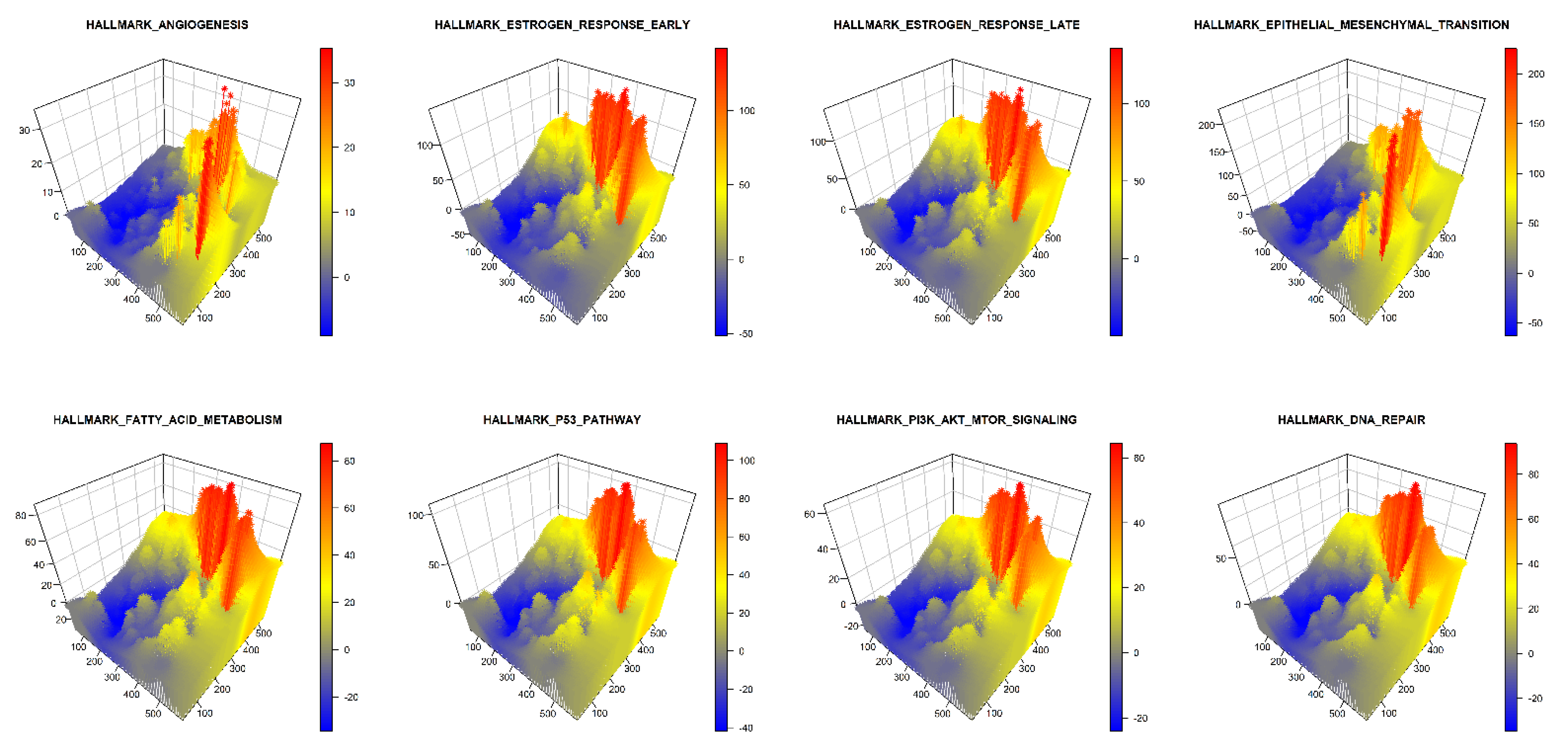
| Gene Sets | Number of Genes in Gene set | Overlap with the Gene List of the Study (%) | Overlap among the 8 Hallmark Gene Sets | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| 1 | HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 201 | 81 (40%) | - | 2% | <1% | <1% | 0 | 0 | <1% | <1% |
| 2 | HALLMARK_ANGIOGENESIS | 37 | 12 (32%) | 2% | - | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | HALLMARK_ESTROGEN_RESPONSE_EARLY | 201 | 64 (32%) | <1% | 0 | - | 8% | <1% | 0 | <1% | <1% |
| 4 | HALLMARK_ESTROGEN_RESPONSE_LATE | 201 | 62 (31%) | 0 | 0 | 8% | - | 0 | 0 | <1% | 1% |
| 5 | HALLMARK_DNA_REPAIR | 151 | 42 (28%) | 0 | 0 | <1% | 0 | - | 0 | 0 | <1% |
| 6 | HALLMARK_PI3K_AKT_MTOR_SIGNALING | 106 | 28 (26%) | 0 | 0 | 0 | 0 | 0 | - | 0 | <1% |
| 7 | HALLMARK_FATTY_ACID_METABOLISM | 159 | 41 (26%) | <1% | 0 | <1% | <1% | 0 | 0 | - | <1% |
| 8 | HALLMARK_P53_PATHWAY | 201 | 50 (25%) | <1% | 0 | <1% | 1% | <1% | <1% | <1% | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajihosseini, M.; Amini, P.; Voicu, D.; Dinu, I.; Pyne, S. Geostatistical Modeling and Heterogeneity Analysis of Tumor Molecular Landscape. Cancers 2022, 14, 5235. https://doi.org/10.3390/cancers14215235
Hajihosseini M, Amini P, Voicu D, Dinu I, Pyne S. Geostatistical Modeling and Heterogeneity Analysis of Tumor Molecular Landscape. Cancers. 2022; 14(21):5235. https://doi.org/10.3390/cancers14215235
Chicago/Turabian StyleHajihosseini, Morteza, Payam Amini, Dan Voicu, Irina Dinu, and Saumyadipta Pyne. 2022. "Geostatistical Modeling and Heterogeneity Analysis of Tumor Molecular Landscape" Cancers 14, no. 21: 5235. https://doi.org/10.3390/cancers14215235
APA StyleHajihosseini, M., Amini, P., Voicu, D., Dinu, I., & Pyne, S. (2022). Geostatistical Modeling and Heterogeneity Analysis of Tumor Molecular Landscape. Cancers, 14(21), 5235. https://doi.org/10.3390/cancers14215235