
Tyrosine Kinase Inhibitor Independent Gene Expression Signature in CML Offers New Targets for LSPC Eradication Therapy

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Code Availability
2.2. Primary Patient Material
2.3. Microarray Analysis
2.4. RNA Sequencing
2.5. Bulk RNA-Seq Analysis
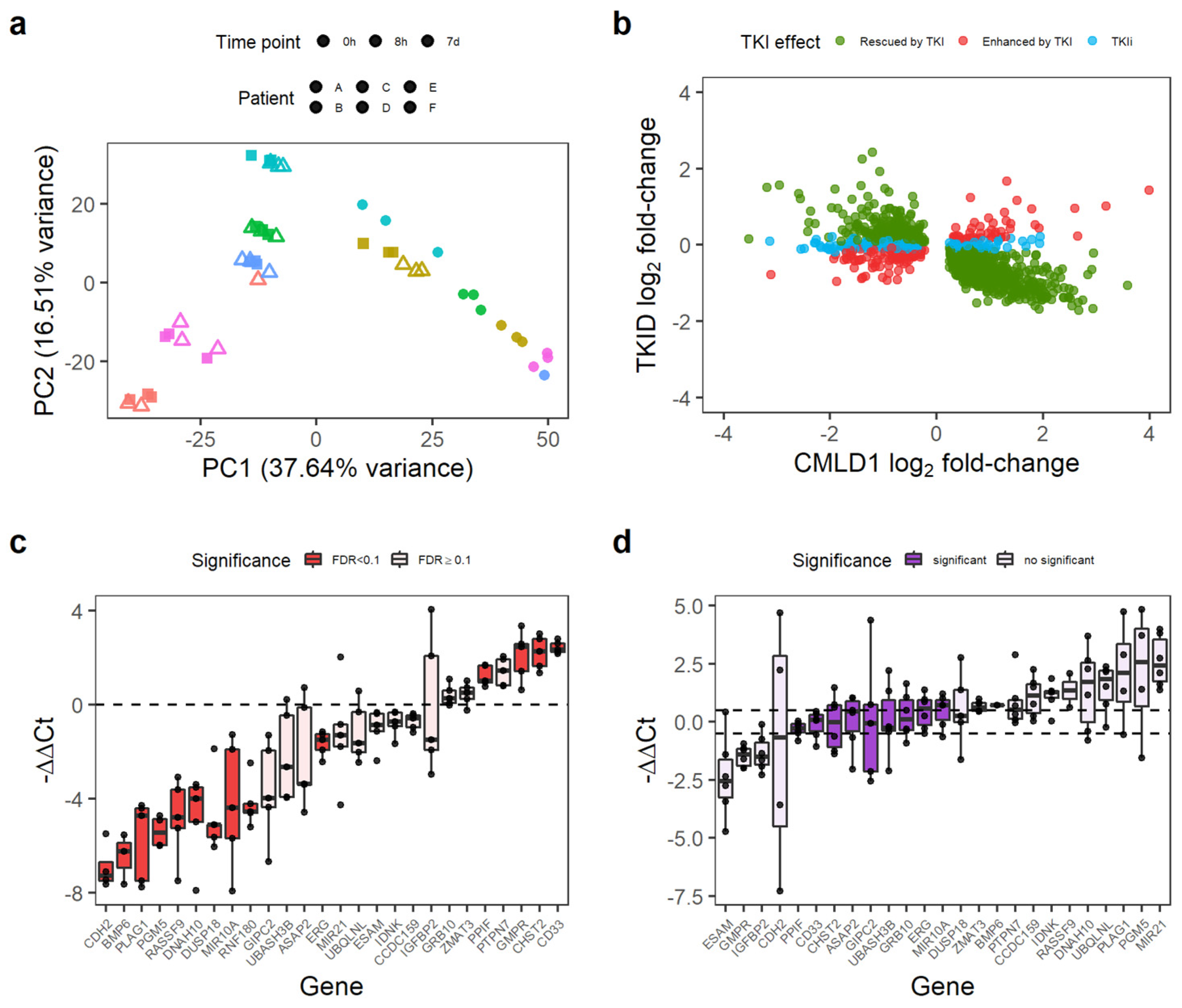
2.6. Validation of the TKI-Independent Signature
2.7. qPCR Analysis
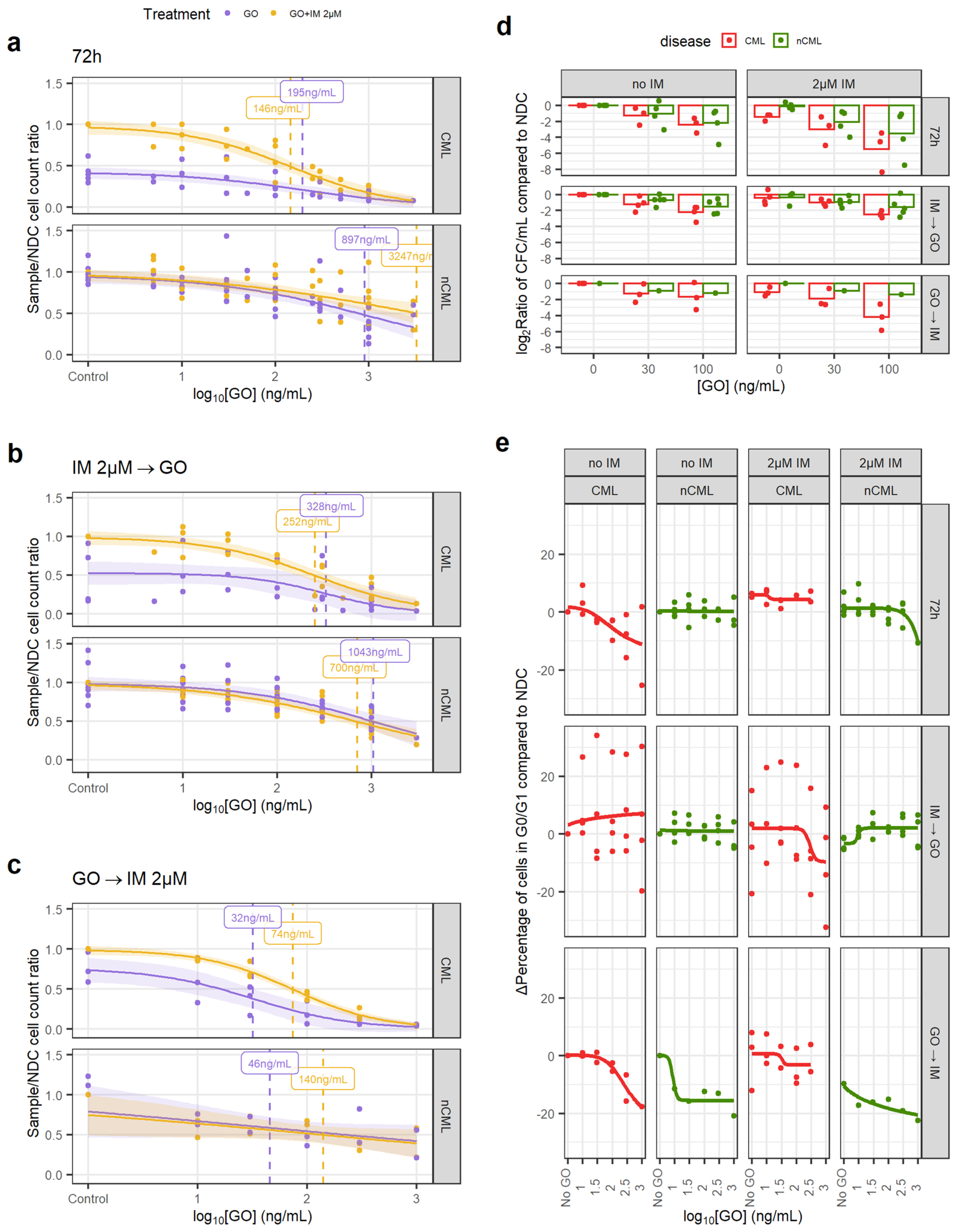
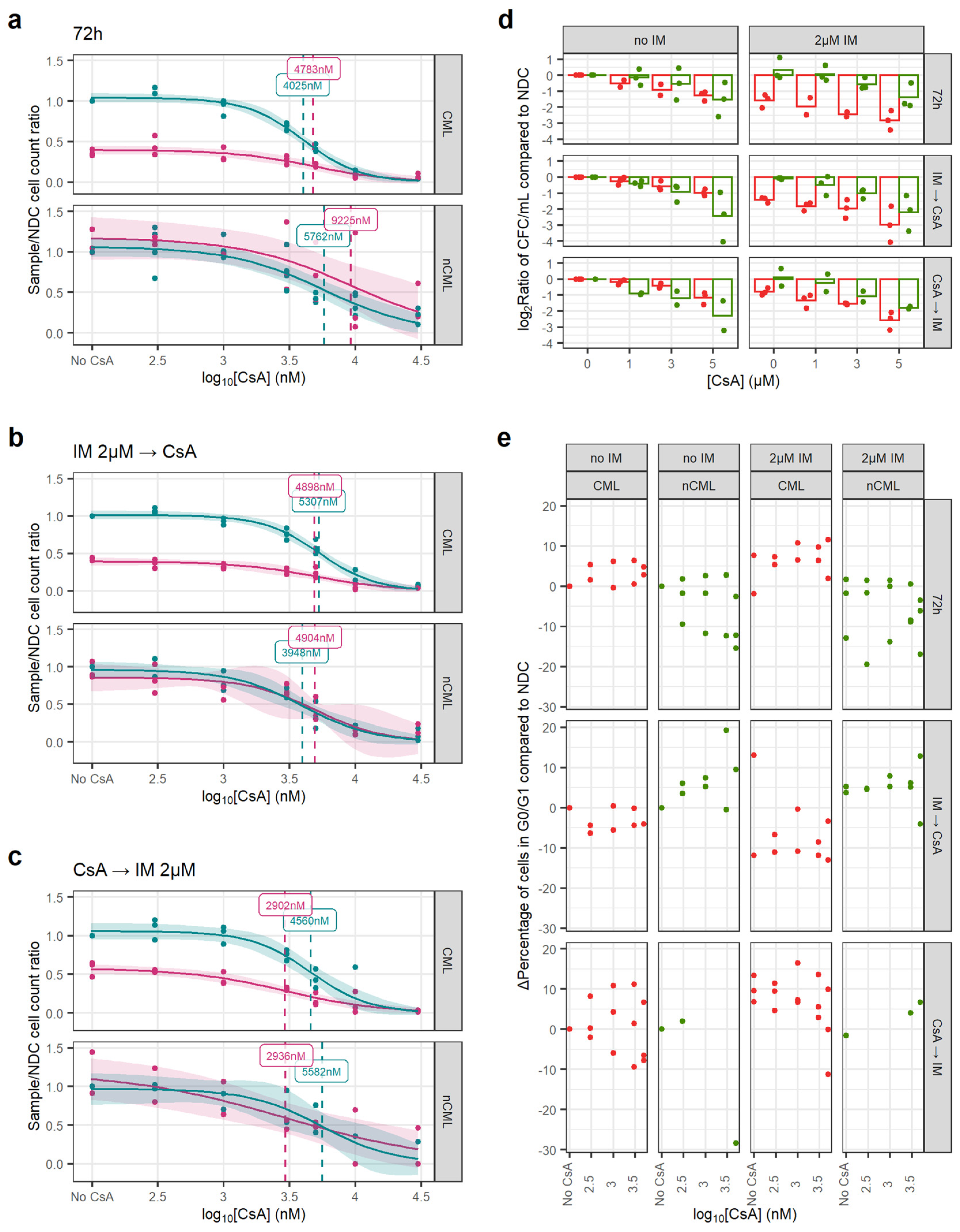
2.8. Drug Response Experiments
- 72 h of GO or CsA with or without IM.
- 72 h IM (or no drug) followed by 72 h of either GO or CsA.
- 72 h of either GO or CsA followed by 72 h of IM (or no drug).
2.9. Colony Forming Cell Assays
2.10. Cell Cycle Analysis
2.11. Other Data Analysis
3. Results
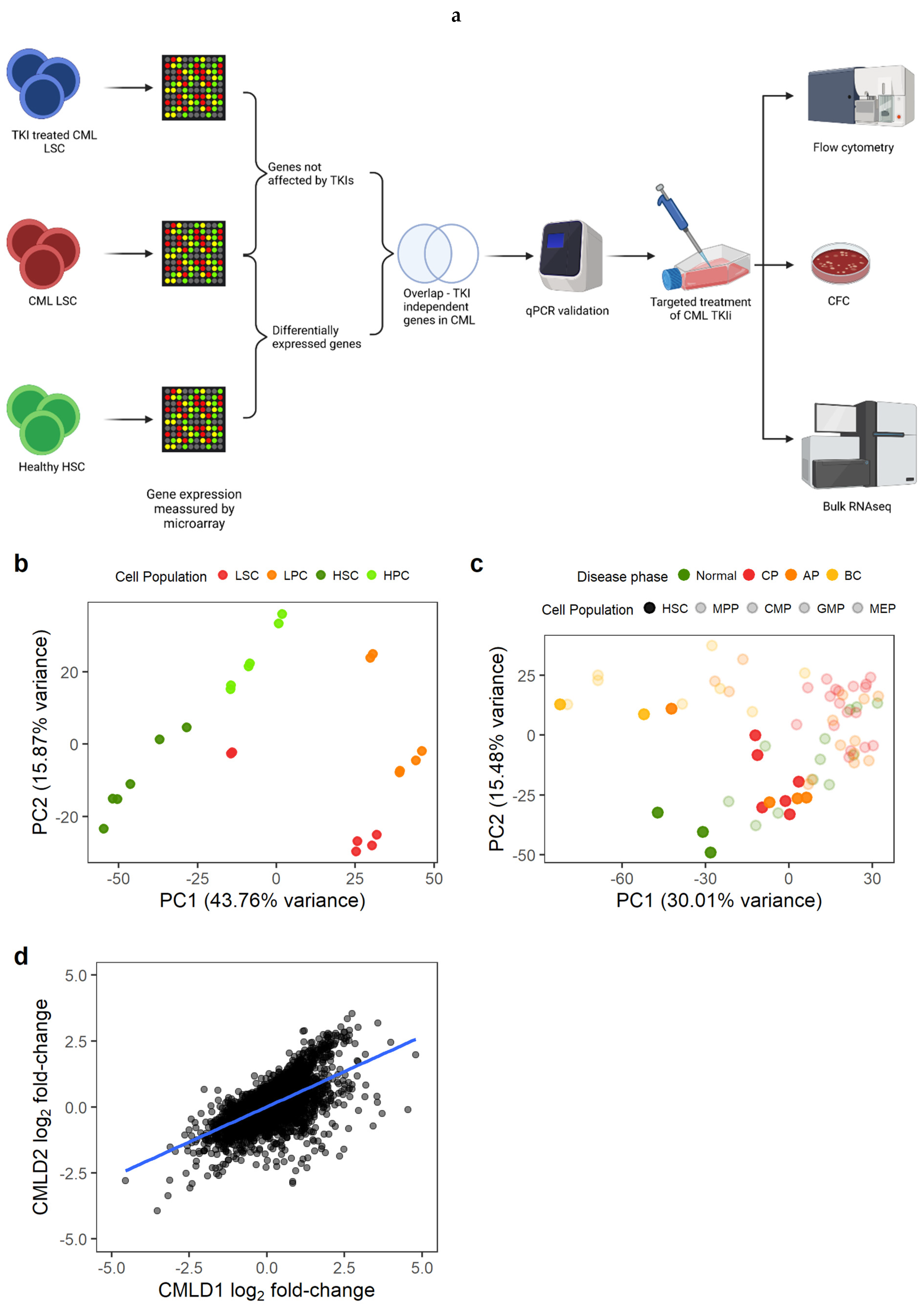
3.1. CML LSC Possess a TKI-Independent Gene Expression Signature
3.2. Anti-CD33 Therapeutic Antibody Can Be Used to Specifically Eradicate CML CD34+ Cells
3.3. CsA Targets CML SPC When in Combination with IM
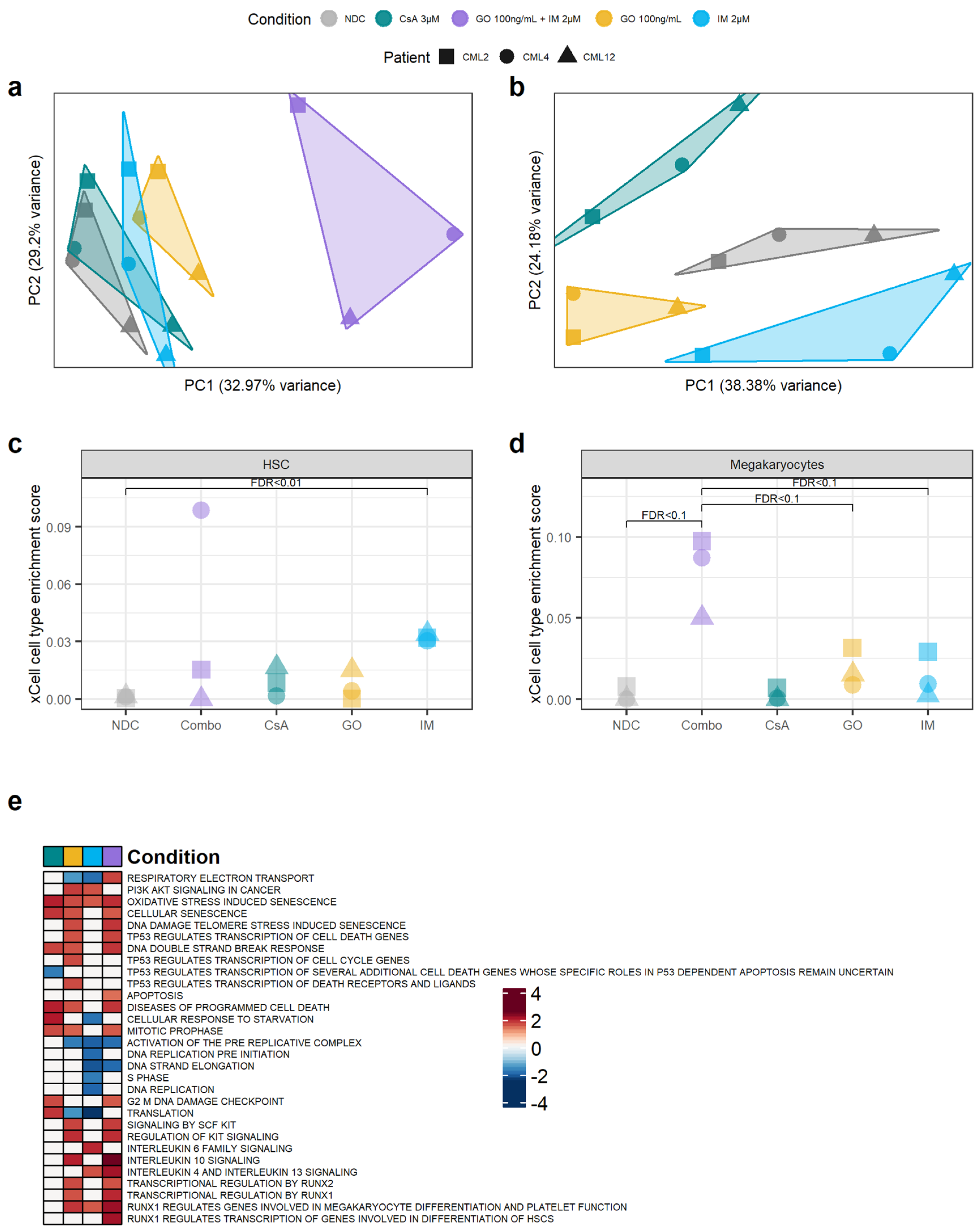
3.4. Transcriptomic Changes after GO, CsA and IM Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, F.X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Charaf, L.; Mahon, F.X.; Lamrissi-Garcia, I.; Moranvillier, I.; Beliveau, F.; Cardinaud, B.; Dabernat, S.; de Verneuil, H.; Moreau-Gaudry, F.; Bedel, A. Effect of tyrosine kinase inhibitors on stemness in normal and chronic myeloid leukemia cells. Leukemia 2016, 31, 65. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.-L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, A.S.; O’Hare, T.; Gu, Z.; Kraft, I.L.; Eiring, A.M.; Khorashad, J.S.; Pomicter, A.D.; Zhang, T.Y.; Eide, C.A.; Manley, P.W.; et al. KIT signaling governs differential sensitivity of mature and primitive CML progenitors to tyrosine kinase inhibitors. Cancer Res. 2013, 73, 5775–5786. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Jimenez-Velasco, A.; San Jose-Eneriz, E.; Garate, L.; Bandres, E.; Cordeu, L.; Aparicio, O.; Saez, B.; Navarro, G.; Vilas-Zornoza, A.; et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol. Cancer Res. 2008, 6, 1830–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiring, A.M.; Khorashad, J.S.; Anderson, D.J.; Yu, F.; Redwine, H.M.; Mason, C.C.; Reynolds, K.R.; Clair, P.M.; Gantz, K.C.; Zhang, T.Y.; et al. beta-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia 2015, 29, 2328–2337. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef] [Green Version]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, K.J.; Rehn, M.; Hasemann, M.S.; Rapin, N.; Bagger, F.O.; Ohlsson, E.; Willer, A.; Frank, A.K.; Sondergaard, E.; Jendholm, J.; et al. ERG promotes the maintenance of hematopoietic stem cells by restricting their differentiation. Genes Dev. 2015, 29, 1915–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CPMP. Points to consider on switching between superiority and non-inferiority. Br. J. Clin. Pharmacol. 2001, 52, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 April 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2016, 10, e0146021. [Google Scholar] [CrossRef] [Green Version]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Irvine, D.; Munje, C.; Castañeda, E.G.; Moka, H.A.; Dunn, K.; Cassels, J.E.; Parry, N.; et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia 2020, 34, 1613–1625. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. 27/11/2020 Mylotarg—EMEA/H/C/004204—N/0020. Available online: https://www.ema.europa.eu/en/documents/product-information/mylotarg-epar-product-information_en.pdf (accessed on 10 September 2021).
- Herrmann, H.; Cerny-Reiterer, S.; Gleixner, K.V.; Blatt, K.; Herndlhofer, S.; Rabitsch, W.; Jager, E.; Mitterbauer-Hohendanner, G.; Streubel, B.; Selzer, E.; et al. CD34(+)/CD38(−) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica 2012, 97, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Jiri, P.; Eugenie, N.; Kamil, K.; Wenda, W. Cyclosporine A: Chemistry and Toxicity—A Review. Curr. Med. Chem. 2021, 28, 3925–3934. [Google Scholar] [CrossRef]
- Grant, D.; Kneteman, N.; Tchervenkov, J.; Roy, A.; Murphy, G.; Tan, A.; Hendricks, L.; Guilbault, N.; Levy, G. Peak cyclosporine levels (cmax) correlate with freedom from liver graft rejection: Results of a Prospective, Randomized Comparison of Neoral and Sandimmune for Liver Transplantation (NOF-8): 1,2. Transplantation 1999, 67, 1133–1137. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Mabbott, N.A.; Baillie, J.K.; Brown, H.; Freeman, T.C.; Hume, D.A. An expression atlas of human primary cells: Inference of gene function from coexpression networks. BMC Genom. 2013, 14, 632. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Garcia-Manero, G.; Cortes, J.; Ravandi, F.; Plunkett, W.; Gandhi, V.; Faderl, S.; O’Brien, S.; Borthakur, G.; Kadia, T.; et al. Twice-Daily Fludarabine and Cytarabine Combination With or Without Gentuzumab Ozogamicin is Effective in Patients with Relapsed/Refractory Acute Myeloid Leukemia, High-Risk Myelodysplastic Syndrome, and Blast-Phase Chronic Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Ovilla-Martinez, R.; Weber Sánchez, L.A.; Cota-Rangel, X.; Baez-Islas, P.E. Gemtuzumab-ozogamicin and blinatumomab as treatment for refractory mixed-phenotype blast crisis in chronic myeloid leukaemia. BMJ Case Rep. 2021, 14, e243745. [Google Scholar] [CrossRef]
- Parsi, M.; Budak-Alpdogan, T. Promyelocytic Blast Crisis of Chronic Myeloid Leukemia in a Patient Undergoing Therapy with a Tyrosine Kinase Inhibitor. Cureus 2020, 12, e7217. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.P.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and Other Kinases from Chronic Myeloproliferative Disorders Stimulate Single-Strand Annealing, an Unfaithful DNA Double-Strand Break Repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef] [Green Version]
- Stoklosa, T.; Poplawski, T.; Koptyra, M.; Nieborowska-Skorska, M.; Basak, G.; Slupianek, A.; Rayevskaya, M.; Seferynska, I.; Herrera, L.; Blasiak, J.; et al. BCR/ABL Inhibits Mismatch Repair to Protect from Apoptosis and Induce Point Mutations. Cancer Res. 2008, 68, 2576–2580. [Google Scholar] [CrossRef] [Green Version]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.-U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef]
- Carreira, R.S.; Lee, Y.; Ghochani, M.; Gustafsson, A.B.; Gottlieb, R.A. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 2010, 6, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem. 2009, 284, 9692–9699. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef] [Green Version]
- Houshmand, M.; Garello, F.; Stefania, R.; Gaidano, V.; Cignetti, A.; Spinelli, M.; Fava, C.; Nikougoftar Zarif, M.; Galimberti, S.; Pungolino, E.; et al. Targeting Chronic Myeloid Leukemia Stem/Progenitor Cells Using Venetoclax-Loaded Immunoliposome. Cancers 2021, 13, 1311. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Castañeda, E.; Hopcroft, L.E.M.; Rogers, S.; Munje, C.; Bittencourt-Silvestre, J.; Copland, M.; Vetrie, D.; Holyoake, T.; Jørgensen, H.G. Tyrosine Kinase Inhibitor Independent Gene Expression Signature in CML Offers New Targets for LSPC Eradication Therapy. Cancers 2022, 14, 5253. https://doi.org/10.3390/cancers14215253
Gómez-Castañeda E, Hopcroft LEM, Rogers S, Munje C, Bittencourt-Silvestre J, Copland M, Vetrie D, Holyoake T, Jørgensen HG. Tyrosine Kinase Inhibitor Independent Gene Expression Signature in CML Offers New Targets for LSPC Eradication Therapy. Cancers. 2022; 14(21):5253. https://doi.org/10.3390/cancers14215253
Chicago/Turabian StyleGómez-Castañeda, Eduardo, Lisa E. M. Hopcroft, Simon Rogers, Chinmay Munje, Joana Bittencourt-Silvestre, Mhairi Copland, David Vetrie, Tessa Holyoake, and Heather G. Jørgensen. 2022. "Tyrosine Kinase Inhibitor Independent Gene Expression Signature in CML Offers New Targets for LSPC Eradication Therapy" Cancers 14, no. 21: 5253. https://doi.org/10.3390/cancers14215253
APA StyleGómez-Castañeda, E., Hopcroft, L. E. M., Rogers, S., Munje, C., Bittencourt-Silvestre, J., Copland, M., Vetrie, D., Holyoake, T., & Jørgensen, H. G. (2022). Tyrosine Kinase Inhibitor Independent Gene Expression Signature in CML Offers New Targets for LSPC Eradication Therapy. Cancers, 14(21), 5253. https://doi.org/10.3390/cancers14215253