
Molecular Events in the Melanogenesis Cascade as Novel Melanoma-Targeted Small Molecules: Principle and Development

 ,
,  ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract
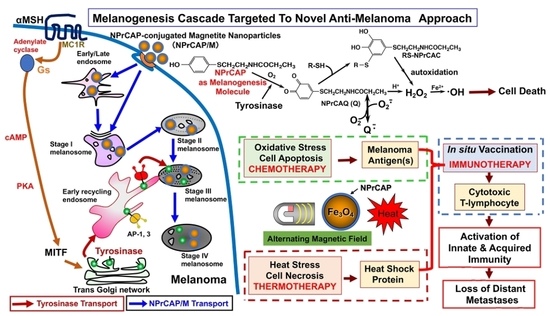
1. Introduction: Overall View of Melanogenesis Cascade to Develop Novel Antimelanoma Approaches by Exploiting Melanogenesis-Based Small Molecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Abbreviation | Structure | Synthesis | Tyrosinase Substrate a | In Vitro Cytotoxicity b | In Vivo Antimelanoma Effect c | In Vivo Depigmentation d |
|---|---|---|---|---|---|---|---|
| 4-S-Cysteinylphenol | 4SCP | 1 | [9] | Yes [16] | [9,21] | Yes/No [9,10,16] | Yes/No [16,28] |
| 2-S-Cysteinylphenol | 2SCP | 2 | [9] | No [16] | [9] | No [16] | |
| 4-S-Cysteinylcatechol | 4SCC | 3 | [9] | Yes [16] | [9,21] | No [9,10] | No [16] |
| 3-S-Cysteinylcatechol | 3SCC | 4 | [9] | [9,21] | No [10] | ||
| 2-S-Cysteinylhydroquinone | 2SCHQ | 5 | [10] | No [16] | [21] | No [10] | No [16,28] |
| 4-S-Cysteaminylphenol | 4SCAP | 6 | [10] | Yes [16,17] | [11,21,22] | Yes [10,16,23,24,25] | Yes [16,23,29] |
| 2-S-Cysteaminylphenol | 2SCAP | 7 | [10] | Yes [16] | No [10] | ||
| 4-S-Homocysteaminylphenol | 4SHCAP | 8 | [11] | Yes [17] | [11] | Yes [29] | |
| 4-S-α-Methylcysteaminylphenol | 4SMeCAP | 9 | [11] | Yes [17] | [11] | Yes [29] | |
| N,N-Dimethylcysteaminylphenol | N,N-DiMeCAP | 10 | [11] | Yes [17] | [11] | Yes [29] | |
| (R)- or (S)-4-S-α-Methylcysteaminylphenol | R,S-4SMeCAP | 11 | [12] | Yes [12] | [12] | Yes [26] | Yes [26] |
| (R)- or (S)-4-S-α-Ethylcysteaminylphenol | R,S-4SEtCAP | 12 | [12] | Yes [12] | [12] | Yes [26] | Yes [26] |
| 4-S-Cysteaminylcatechol | 4SCAC | 13 | [13] | Yes [13] | [13] | Yes [13] | |
| 3-S-Cysteaminylcatechol | 3SCAC | 14 | [13] | No [13] | [13] | No [13] | |
| 2-S-Cysteiaminylhydroquinone | 2SCAHQ | 15 | [13] | No [13] | [13] | No [13] | |
| N-Acetyl-4-S-cysteaminylphenol | NAcCAP | 16 | [14] | Yes [18] | [19] | Yes [24] | Yes [29,30] |
| N-Propionyl-4-S-cysteaminylphenol | NPrCAP | 17 | [15] | Yes [15,18,20] | [15,18,19] | Yes [15,27] | Yes [15,18] |

2. Advances in Anti-Melanoma Targeted Small Molecules and Mechanisms in Relation to the Melanogenesis Cascade
2.1. Chemotherapeutic Approaches Using the Initial Step of Melanogenesis: Tyrosine, Dopa and Their Analogues
- (a)
- Tyrosine
- (b) L-Dopa/dopamine and related analogues
- (c) Sulfur homologues of tyrosine and related compounds
2.2. Mechanism of Anti-Melanoma Action in Relation to the Melanogenesis Cascade
3. Thermal Medicine for Selective Anti-Melanoma Therapy Utilizing Melanogenesis Small Molecules
3.1. Melanogenesis Molecule-Based Boron Neutron Thermal Medicine: Reaction of Thermal Neutrons with Boron 10 Conjugated to Dopa Analogue Para-Boronophenylalanine Hydrochloride (10B1-BPA)
- (a)
- Principle and pharmacokinetics of 10B1-BPA Thermal Neutron Capture therapy
- (b) In vivo radiotherapeutic studies and preclinical experiments
- (c) Clinical trial of melanoma patients using 10B1-BPA thermal neutron capture therapy (Boron Neutron Capture Therapy, BNCT)
3.2. Melanogenesis-Based Antimelanoma Thermal Medicine by Conjugation with Magnetite Nanoparticles; Establishment of Melanoma Chemo-Thermo-Immunotherapy (CTI Therapy)
- (a)
- Principle of magnetite hyperthermia for cancer nanomedicine
- (b) Conjugation of melanogenesis molecule NPrCAP with magnetite nanoparticles for novel antimelanoma thermotherapy
- (c) Development of the chemo-thermo-immuno-therapy approach
- (d) Immunological effects of melanogenesis-targeted anti-melanoma molecules; orchestration of innate and adaptive immunity by CTI therapy
- (e) Preliminary clinical trials for human melanoma patients
4. Anti-Melanoma Approach for Unresectable/Metastatic Melanoma Patients by Currently Available Targeted Therapies and Immune Checkpoint Inhibitors
4.1. Ipilimumab
4.2. Anti-PD-1 Antibodies: Nivolumab and Pembrolizumab
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMF | Alternating magnetic field |
| AP | Activator protein |
| APC | Antigen presenting cells |
| APTES | 3-Aminopropyltriethoxysilane |
| 10B1 | Boron-10 |
| 10B1-BPA | 10B1-para-Boronophenylalanine hydrochloride |
| BNCTT | Boron neutron capture thermal therapy |
| BRAFi | Braf inhibitor |
| CAP | Cysteaminylphenol |
| CP | Cysteinylphenol |
| CTI therapy | Chemo-thermo-immunotherapy |
| CTL | Cytotoxic T lymphocytes |
| CTLA-4 | Cytotoxic T lymphocyte-associated 4 |
| DCs | Dendric cells |
| Dopeba | 3,4-dihydroxyphenethylboric acid |
| DDS | Drug delivery system |
| GSH | Glutathione |
| HSPs | Heat shock proteins |
| ICI | Immune checkpoint inhibitor |
| ip | Intraperitoneal injection |
| irAE | Immune-related adverse events |
| iv | intravenous infusion |
| M | Magnetite nanoparticle |
| MCL | Magnetite cationic liposome |
| MEKi | MEK inhibitor |
| NAcCAP | N-Acetyl-4SCAP |
| NPrCAP | N-Propionyl-4-S-CAP |
| NPrCAQ | N-Propionyl-4-S-cysteaminyl-1,2-benzoquinone |
| OS | Overall survival |
| PD-1 | Programmed cell death-1 |
| PEG | Polyethylene glycol |
| RFS | Recurrence-free survival |
| ROS | Reactive oxygen species |
| 4SCAC | 4-S-Cysteaminylcatechol |
| 4SCAP | 4-S-Cysteaminylphenol |
| 5SCD | 5-S-Cysteinyldopa |
| 4SCP | 4-S-Cysteinylphenol |
| TGN | Trans-Golgi network |
| TLR | Toll-like receptors |
References
- Jimbow, K.; Fitzpatrick, T.B.; Quevedo, W.C., Jr. Formation, chemical compositions and functions of melanin pigments in mammals. Biol. Integument 1986, 2, 278–292. [Google Scholar] [CrossRef]
- Wick, M. An experimental approach to the chemotherapy of melanoma. J. Investig. Dermatol. 1980, 74, 63–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The toxicity of melanin precursors. J. Investig. Dermatol. 1978, 70, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbow, K.; Iwashina, T.; Alena, F.; Yamada, K.; Pankovich, J.; Umemura, T. Exploitation of pigment biosynthesis pathway as a selective chemotherapeutic approach for malignant melanoma. J. Investig. Dermatol. 1993, 100, 231S–238S. [Google Scholar] [CrossRef] [Green Version]
- Prota, G. Melanins and Melanogenesis; Academic Press: San Diego, CA, USA, 1992; pp. 1–290. [Google Scholar]
- Prota, G.; d’Ischia, M.; Mascagna, D. Melanogenesis as a targeting strategy against metastatic melanoma: A reassessment. Melanoma Res. 1994, 4, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.A.; Cooksey, C.J.; Johnson, C.I.; Land, E.J.; Latter, A.M.; Ramsden, C.A. Melanogenesis-targeted anti-melanoma pro-drug development: Effect of side chain variations on the cytotoxicity of tyrosinase-generated ortho-quinones in a model screening system. Eur. J. Cancer 1997, 33, 135–143. [Google Scholar] [CrossRef]
- Ito, S.; Sugumaran, M.; Wakamatsu, K. Chemical reactivities of ortho-quinones produced in living organisms: Fate of quinonoid products formed by tyrosinase and phenoloxidase action on phenols and catechols. Int. J. Mol. Sci. 2020, 21, 6080. [Google Scholar] [CrossRef]
- Ito, S.; Inoue, S.; Yamamoto, Y.; Fujita, K. Synthesis and antitumor activity of cysteinyl-3,4-dihydroxyphenylalanines and related compounds. J. Med. Chem. 1981, 24, 673–677. [Google Scholar] [CrossRef]
- Miura, S.; Ueda, T.; Jimbow, K.; Ito, S.; Fujita, K. Synthesis of cysteinylphenol, cysteaminylphenol and related compounds and in vivo evaluation of anti-melanoma effect. Arch. Dermatol. Res. 1987, 279, 219–225. [Google Scholar] [CrossRef]
- Inoue, S.; Ito, S.; Wakamatsu, K.; Jimbow, K.; Fujita, K. Mechanism of growth inhibition of melanoma cells by 4-S-cysteaminylphenol and its analogues. Biochem. Pharmacol. 1990, 39, 1077–1083. [Google Scholar] [CrossRef]
- Yukitake, J.; Otake, H.; Inoue, S.; Wakamatsu, K.; Olivares, C.; Solano, F.; Hasegawa, K.; Ito, S. Synthesis and selective in vitro anti-melanoma effect of enantiomeric alpha-methyl- and alpha-ethyl-4-S-cysteaminylphenol. Melanoma Res. 2003, 13, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Hasegawa, K.; Ito, S.; Ozeki, H.; Solano, F.; Jiménez-Cervantes, C.; Wakamatsu, K.; Fujita, K. Antimelanoma effect of 4-S-cysteaminylcatechol, an activated form of 4-S-cysteaminylphenol. Cancer Res. 1995, 55, 2603–2607. [Google Scholar] [PubMed]
- Padgette, S.R.; Herman, H.H.; Han, J.H.; Pollock, S.H.; May, S.W. Antihypertensive activities of phenyl aminoethyl sulfides, a class of synthetic substrates for dopamine hydroxylase. J. Med. Chem. 1984, 27, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Thomas, P.D.; Shokravi, M.; Singh, S.; Samra, S.; Chang, D.; Jimbow, K. Synthesis and antitumor effect of the melanogenesis-based antimelanoma agent N-propionyl-4-S-cysteaminylphenol. Biochem. Pharmacol. 1998, 55, 2023–2029. [Google Scholar] [CrossRef]
- Jimbow, K.; Miura, T.; Ito, S.; Ishikawa, K. Phenolic melanin precursors provide a rational approach to the design of antitumor agents for melanoma. Pigment Cell Res. 1989, 2, 34–39. [Google Scholar] [CrossRef]
- Pankovich, J.M.; Jimbow, K.; Ito, S. 4-S-cysteaminylphenol and its analogues as substrates for tyrosinase and monoamine oxidase. Pigment Cell Res. 1990, 3, 146–149. [Google Scholar] [CrossRef]
- Minamitsuji, Y.; Toyofuku, K.; Sugiyama, S.; Yamada, K.; Jimbow, K. Sulfur containing tyrosine analogs can cause selective melanocytotoxocity involving tyrosinase-mediated apoptosis. J. Investig. Dermatol. Symp. Proc. 1999, 4, 130–136. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kishi, H.; Cao, H.; Ota, M.; Yamashita, T.; Singh, S.; Jimbow, K. Selective incorporation and specific cytocidal effect as the cellular basis for the antimelanoma action of sulphur containing tyrosine analogs. J. Investig. Dermatol. 1999, 113, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, E.; et al. Mechanism of putative neo-antigen formation from N-propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653. [Google Scholar] [CrossRef]
- Yamada, I.; Seki, S.; Matsubara, O.; Ito, S.; Suzuki, S.; Kasuga, T. The cytotoxicity of cysteinylcatechols and related compounds to human melanoma cells in vitro. J. Investig. Dermatol. 1987, 88, 538–540. [Google Scholar] [CrossRef]
- Yamada, I.; Seki, S.; Ito, S.; Suzuki, S.; Matsubara, O.; Kasuga, T. The killing effect of 4-S-cysteaminylphenol, a newly synthesised melanin precursor, on B16 melanoma cell lines. Br. J. Cancer 1991, 63, 187–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Jimbow, K. Selective cytotoxicity of 4-S-cysteaminylphenol on follicular melanocytes of the black mouse: Rational basis for its application to melanoma chemotherapy. Cancer Res. 1987, 47, 3278–3284. [Google Scholar] [PubMed]
- Miura, T.; Jimbow, K.; Ito, S. The in vivo antimelanoma effect of 4-S-cysteaminylphenol and its N-acetyl derivative. Int. J. Cancer 1990, 46, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Nemoto, T.; Seki, S.; Ito, S.; Kasuga, T. In vivo antimelanoma effects of 4-S-cysteaminylphenol, a newly synthesized therapeutic agent specific to melanoma. J. Cancer Res. Clin. Oncol. 1993, 119, 470–474. [Google Scholar] [CrossRef]
- Yukitake, J.; Otake, H.; Inoue, S.; Wakamatsu, K.; Ito, S. Comparison of in vivo anti-melanoma effect of enantiomeric alpha-methyl- and alpha-ethyl-4-S-cysteaminylphenol. Melanoma Res. 2004, 14, 116–120. [Google Scholar] [CrossRef]
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayama, E.; Okura, M.; et al. N-propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60. [Google Scholar] [CrossRef]
- Ito, Y.; Jimbow, K.; Ito, S. Depigmentation of black guinea pig skin by topical application of cysteaminylphenol, cysteinylphenol, and related compounds. J. Investig. Dermatol. 1987, 88, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Alena, F.; Jimbow, K.; Ito, S. Melanocytotoxicity and antimelanoma effects of phenolic amine compounds in mice in vivo. Cancer Res. 1990, 50, 3743–3747. [Google Scholar] [PubMed]
- Wong, M.; Jimbow, K. Selective cytotoxicity of N-acetyl-4-S-cysteaminylphenol on follicular melanocytes of black mice. Brit. J. Dermatol. 1991, 124, 56–61. [Google Scholar] [CrossRef]
- Konno, A.; Sato, N.; Yagihashi, A.; Torigoe, T.; Cho, J.M.; Torimoto, K.; Hara, I.; Wada, Y.; Okubo, M.; Takahashi, N. Heat- or stress-inducible transformation-associated cell surface antigen on the activated H-ras oncogene-transfected rat fibroblast. Cancer Res. 1989, 49, 6578–6582. [Google Scholar] [PubMed]
- Ménoret, A.; Chandawarkar, R. Heat-shock protein-based anticancer immunotherapy: An idea whose time has come. Semin. Oncol. 1998, 25, 654–660. [Google Scholar] [PubMed]
- Srivastava, P.K.; Ménoret, A.; Basu, S.; Binder, R.J.; McQuade, K.L. Heat shock proteins come of age: Primitive functions acquire new roles in an adaptive world. Immunity 1998, 8, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Tsuboi, N.; Sato, N.; Kikuchi, K. 70 kDa heat shock cognate protein is a transformation-associated antigen and a possible target for the host’s anti-tumor immunity. J. Immunol. 1993, 151, 5516–5524. [Google Scholar] [PubMed]
- Tamura, Y.; Peng, P.; Liu, K.; Daou, M.; Srivastava, P.K. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 1997, 278, 117–120. [Google Scholar] [CrossRef]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Denis-Larose, C.; Massie, B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol. 1997, 17, 5317–5327. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Sato, N. Heat shock proteins: Chaperoning of innate and adaptive immunities. Jpn. J. Hyperthermic Oncol. 2003, 19, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, P.K. Immunotherapy for human cancer using heat shock protein-peptide complexes. Curr. Oncol. Rep. 2005, 7, 104–108. [Google Scholar] [CrossRef]
- Takashima, S.; Sato, N.; Kishi, A.; Tamura, Y.; Hirai, I.; Torigoe, T.; Yagihashi, A.; Takahashi, S.; Sagae, S.; Kudo, R.; et al. Involvement of peptide antigens in the cytotoxicity between 70-kDa heat shock cognate protein-like molecule and CD3+, CD4−, CD8−, TCR-αβ- killer T cells. J. Immunol. 1996, 157, 3391–3395. [Google Scholar] [PubMed]
- Yanase, M.; Shinkai, M.; Honda, H.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Antitumor immunity induction by intracellular hyperthermia using magnetite cationic liposomes. Jpn. J. Cancer Res. 1998, 89, 775–782. [Google Scholar] [CrossRef]
- Ueda, G.; Tamura, Y.; Hirai, I.; Kamiguchi, K.; Ichimiya, S.; Torigoe, T.; Hiratsuka, H.; Sunakawa, H.; Sato, N. Tumor-derived heat shock protein 70-pulsed dendritic cells elicit-tumor-specific cytotoxic T lymphocytes (CTLs) and tumor immunity. Cancer Sci. 2004, 95, 248–253. [Google Scholar] [CrossRef]
- Ito, A.; Honda, H.; Kobayashi, T. Cancer immunotherapy based on intracellular hyperthermia using magnetite nanoparticles: A novel concept of “heat-controlled necrosis” with heat shock protein expression. Cancer Immunol. Immunother. 2006, 55, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Cao, T.; Connolly, J.E.; Monnet, L.; Bennett, L.; Chapel, S.; Bagnis, C.; Mannoni, P.; Davoust, J.; Palucka, A.K.; et al. Hyperthermia enhances CTL cross-priming. J. Immunol. 2006, 176, 2134–2141. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Fujioka, M.; Yoshida, T.; Wakamatsu, K.; Ito, S.; Yamashita, T.; Jimbow, K.; Honda, H. 4-S-Cysteaminylphenol-loaded magnetite cationic liposomes for combination therapy of hyperthermia with chemotherapy against malignant melanoma. Cancer Sci. 2007, 98, 424–430. [Google Scholar] [CrossRef]
- Jimbow, K.; Takada, T.; Osai, Y.; Thomas, P.D.; Sato, M.; Sato, A.; Kamiya, T.; Ono, I.; Tamura, Y.; Sato, N.; et al. Melanogenesis exploitation and melanoma nanomedicine: Utilization of melanogenesis substrate, NPrCAP for exploiting melanoma-targeting drug and its conjugation with magnetite nanoparticles for developing melanoma chemo-thermo-immunotherapy. Open Conf. Proc. J. 2011, 2, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Jimbow, K.; Tamura, Y.; Yoneta, A.; Kamiya, T.; Ono, I.; Yamashita, T.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; et al. Conjugation of magnetic nanoparticles with melanogenesis substrate, NPrCAP provides melanoma targeted, in situ peptide vaccine immunotherapy through HSP production by chemo-thermotherapy. J. Biomater. Nanobiotechnol. 2012, 3, 140–153. [Google Scholar] [CrossRef]
- Mizote, Y.; Wakamatsu, K.; Ito, S.; Uenaka, A.; Ohue, Y.; Kurose, K.; Isobe, M.; Ito, A.; Tamura, Y.; Honda, H.; et al. TLR4 and NLRP3 inflammasome activation in monocytes by N-propionyl cysteaminylphenol-maleimide-dextran (NPCMD). J. Dermatol. Sci. 2014, 73, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Ito, A.; Wakamatsu, K.; Kamiya, T.; Torigoe, T.; Honda, H.; Yamashita, T.; Uhara, H.; Ito, S.; Jimbow, K. Immunomodulation of melanoma by chemo-thermo-immunotherapy using conjugates of melanogenesis substrate NPrCAP and magnetite nanoparticles: A review. Int, J. Mol. Sci. 2022, 23, 6457. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Lerner, A.B. 5,6-Dihydroxyindole is a melanin precursor showing potent cytotoxicity. Nature 1978, 276, 626–628. [Google Scholar] [CrossRef]
- Kreider, J.W.; Wade, D.R.; Rosenthal, M.; Densley, T. Maturation and differentiation of B16 melanoma cells induced by theophylline treatment. J. Natl. Cancer Inst. 1975, 54, 1457–1467. [Google Scholar] [CrossRef]
- Pawelek, J.; Wong, G.; Sansone, M.; Morowitz, J. Molecular biology of pigment cells. Molecular controls in mammalian pigmentation. Yale J. Biol. Med. 1973, 46, 430–443. [Google Scholar] [PubMed]
- Wick, M.M.; Kramer, R.A.; Gorman, M. Enhancement of L-dopa incorporation into melanoma by dopa decarboxylase inhibition. J. Investig. Dermatol. 1978, 70, 358–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, M.M.; Frei, E., 3rd. Selective incorporation of L-3,4-dihydroxyphenylalanine by S-91 Cloudman melanoma in vitro. Cancer Res. 1977, 37, 2123–2125. [Google Scholar] [PubMed]
- Wick, M.M. L-Dopa methyl ester as a new antitumour agent. Nature 1977, 269, 512–513. [Google Scholar] [CrossRef] [PubMed]
- Wick, M.M. Dopamine: A novel antitumor agent active against B-16 melanoma in vivo. J. Investig. Dermatol. 1978, 71, 163–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, M.M. The chemotherapy of malignant melanoma. J. Investig. Dermatol. 1983, 80 (Suppl. S1), 61s–62s. [Google Scholar] [CrossRef]
- Wick, M.M.; Byers, L.; Ratliff, J. Selective toxicity of 6-hydroxydopa for melanoma cells. J. Investig. Dermatol. 1977, 72, 67–69. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Ito, S.; Inoue, S.; Yamamoto, Y.; Takeuchi, J.; Shamoto, M.; Nagatsu, T. Selective toxicity of 5-S-cysteinyldopa, a melanin precursor, to tumor cells in vitro and in vivo. Cancer Res. 1980, 40, 2543–2546. [Google Scholar] [PubMed]
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653. [Google Scholar] [PubMed]
- Ito, S.; Inoue, S.; Fujita, K. The mechanism of toxicity of 5-S-cysteinyldopa to tumour cells. Hydrogen peroxide as a mediator of cytotoxicity. Biochem. Pharmacol. 1983, 32, 2079–2081. [Google Scholar] [CrossRef]
- Pankovich, J.M.; Jimbow, K. Tyrosine transport in a human melanoma cell line as a basis for selective transport of cytotoxic analogues. Biochem. J. 1991, 280, 721–725. [Google Scholar] [CrossRef]
- Nakamura, T.; Seki, S.; Matsubara, O.; Ito, S.; Kasuga, T. Specific incorporation of 4-S-cysteinylphenol into human melanoma cells. J. Investig. Dermatol. 1988, 90, 725–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.-Y.; Lin, C.-C.; Hsieh, Y.-S.; Wu, Y.-T. Nanoformulation development to improve the biopharmaceutical properties of fisetin using design of experiment approach. Molecules 2021, 26, 3031. [Google Scholar] [CrossRef] [PubMed]
- Lant, N.J.; McKeown, P.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and antimelanoma activity of analogues of N-acetyl-4-S-cysteaminylphenol. Anticancer. Drug Des. 2000, 15, 295–302. [Google Scholar] [PubMed]
- Lant, N.J.; McKeown, P.; Timoney, M.C.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and anti-melanoma activity of analogues of N-acetyl-4-S-cysteaminylphenol substituted with two methyl groups alpha to the nitrogen. Anticancer. Drug Des. 2001, 6, 49–55. [Google Scholar] [PubMed]
- Pearson, V.C.; Ferguson, J.; Rogers, P.M.; Kelland, L.R.; Robins, D.J. Synthesis and antimelanoma activity of tertiary amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2003, 13, 503–512. [Google Scholar] [CrossRef]
- Ferguson, J.; Rogers, P.M.; Kelland, L.R.; Robins, D.J. Synthesis and antimelanoma activity of sterically congested tertiary amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2005, 15, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, K.; Robertson, J.; Lant, N.; Kelland, L.R.; Rogers, P.M.; Robins, D.J. Synthesis and antimelanoma activity of reversed amide analogues of N-acetyl-4-S-cysteaminylphenol. Oncol. Res. 2006, 16, 97–106. [Google Scholar] [CrossRef]
- Granada, M.; Mendes, E.; Perry, M.J.; Penetra, M.J.; Gaspar, M.M.; Pinho, J.O.; Serra, S.; António, C.T.; Francisco, A.P. Sulfur Analogues of Tyrosine in the Development of Triazene Hybrid Compounds: A New Strategy against Melanoma. ACS Med. Chem. Lett. 2021, 12, 1669–1677. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef]
- Tse, D.C.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40. [Google Scholar] [CrossRef]
- Tanaka, H.; Yamashita, Y.; Umezawa, K.; Hirobe, T.; Ito, S.; Wakamatsu, K. The pro-oxidant activity of pheomelanin is significantly enhanced by UVA irradiation: Benzothiazole moieties are more reactive than benzothiazine moieties. Int. J. Mol. Sci. 2018, 19, 2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alena, F.; Dixon, W.; Thomas, P.; Jimbow, K. Glutathione plays a key role in the depigmenting and melanocytotoxic action of N-acetyl-4-S-cysteaminylphenol in black and yellow hair follicles. J. Investig. Dermatol. 1995, 104, 792–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alena, F.; Iwashina, T.; Gili, A.; Jimbow, K. Selective in vivo accumulation of N-acetyl-4-S-cysteaminylphenol in B16F10 murine melanoma and enhancement of its in vitro and in vivo antimelanoma effect by combination of buthionine sulfoximine. Cancer Res. 1994, 54, 2661–2666. [Google Scholar] [PubMed]
- Ito, S.; Kato, T.; Fujita, K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem. Pharmacol. 1988, 37, 1707–1710. [Google Scholar] [CrossRef]
- Prezioso, J.A.; Wang, N.; Bloomer, W.D. Thymidylate synthase as a target enzyme for the melanoma-specific toxicity of 4-S-cysteaminylphenol and N-acetyl-4-S-cysteaminylphenol. Cancer Chemother. Pharmacol. 1992, 30, 394–400. [Google Scholar] [CrossRef]
- Ito, S.; Kato, T.; Ishikawa, K.; Kasuga, T.; Jimbow, K. Mechanism of selective toxicity of 4-S-cysteinylphenol and 4-S-cysteaminylphenol to melanocytes. Biochem. Pharmacol. 1987, 36, 2007–2011. [Google Scholar] [CrossRef]
- Penning, T.M. Genotoxicity of ortho-quinones: Reactive oxygen species versus covalent modification. Toxicol. Res. 2017, 6, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Dunsmore, L.; Navo, C.D.; Becher, J.; de Montes, E.G.; Guerreiro, A.; Hoyt, E.; Brown, L.; Zelenay, V.; Mikutis, S.; Copper, J.; et al. Controlled masking and targeted release of redox-cycling ortho-quinones via a C-C bond-cleaving 1,6-elimination. Nat. Chem. 2022, 14, 754–765. [Google Scholar] [CrossRef]
- Gutierrez, P.L. The metabolism of quinone-containing alkylating agents: Free radical production and measurement. Front. Biosci. 2000, 5, D629–D638. [Google Scholar] [CrossRef] [Green Version]
- Mascagna, D.; Costantini, C.; d’Ischia, M.; Prota, G. Biomimetic oxidation of the antimelanoma agent 4-S-cysteaminylphenol and related catechol thioethers: Isolation and reaction behaviour of novel dihydrobenzothiazinequinones. Tetrahedron 1994, 50, 8757–8764. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-1,4-benzothiazine-6,7-dione, the ultimate toxic metabolite of 4-S-cysteaminylphenol and 4-S-cysteaminylcatechol. Biochem. Pharmacol. 1997, 53, 1435–1444. [Google Scholar] [CrossRef]
- Mishima, Y.; Kondoh, H. Dual control of melanogenesis and melanoma growth: Overview molecular to clinical level and the reverse. Pigment Cell Res. 2000, 13 (Suppl. S8), 10–22. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Ichihashi, M.; Tsuji, M.; Hatta, S.; Ueda, M.; Honda, C.; Suzuki, T. Treatment of malignant melanoma by selective thermal neutron capture therapy using melanoma-seeking compound. J. Investig. Dermatol. 1989, 92 (Suppl. S5), 321S–325S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, M.; Ichihashi, M.; Mishima, Y. Selective affinity of 10B1-paraboronophenylalanine-HCl to malignant melanoma: Thermal neutron capture therapy. Hihon Hifuka Gakkai Zasshi 1983, 93, 773–778. [Google Scholar] [PubMed]
- Takagaki, M.; Mishima, Y.; Ichihashi, M.; Fukuda, H.; Matsuzawa, T. Imaging of Boron-10 distribution in tissue and its quantitative analysis using α-track autoradiography. KURRI-TR 1985, 260, 69–72. [Google Scholar]
- Tamauchi, H.; Tamaoki, N.; Ueda, M.; Mishima, Y. Therapeutic model in nude mice carrying transplanted human melanoma line: Studies of selective thermal neutron capture therapy. KURRI-TR 1985, 260, 91–93. [Google Scholar]
- Karashima, H.; Hiratsuka, J.; Mishima, Y. Clinical dosimetry using human phantom with skeletal bone for thermal neutron capture therapy. KURRI-TR 1985, 260, 63–67. [Google Scholar]
- Mishima, Y.; Ichihashi, M.; Hatta, S.; Honda, C.; Yamamura, K.; Nakagawa, T. New thermal neutron capture therapy for malignant melanoma: Melanogenesis-seeking 10B molecule-melanoma cell interaction from in vitro to first clinical trial. Pigment Cell Res. 1989, 2, 226–234. [Google Scholar] [CrossRef]
- Fukuda, H. Boron neutron capture therapy (BNCT) for cutaneous malignant melanoma using 10B-p-boronophenylalanine (BPA) with special reference to the radiobiological basis and clinical results. Cells 2021, 10, 2881. [Google Scholar] [CrossRef]
- Fukuda, H.; Hiratsuka, J.; Kobayashi, T.; Sakurai, Y.; Yoshino, K.; Karashima, H.; Turu, K.; Araki, K.; Mishima, Y.; Ichihashi, M. Boron neutron capture therapy (BNCT) for malignant melanoma with special reference to absorbed doses to the normal skin and tumor. Australas. Phys. Eng. Sci. Med. 2003, 26, 97–103. [Google Scholar] [CrossRef]
- Busse, P.M.; Zamenhof, R.; Madoc-Jones, H.; Solares, G.; Kiger, S.; Reley, K.; Chung, C.; Roger, G.; Harling, O. Clinical follow-up of patients with melanoma of the extremity treated in a phase I boron neutron capture therapy protocol. In Advances in Neutron Capture Therapy; Larsson, B., Crawford, J., Weinreich, R., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 1, pp. 60–64. ISBN 0-444-82781-1. [Google Scholar]
- Menéndez, P.R.; Roth, B.M.C.; Pereira, M.D.; Casal, M.R.; González, S.J.; Feld, D.B.; Santa Cruz, G.A.; Kessler, J.; Longhino, J.; Blaumann, H.; et al. BNCT for skin melanoma in extremities: Updated Argentine clinical results. Appl. Radiat. Isot 2009, 67 (Suppl. S7–S8), s50–s53. [Google Scholar] [CrossRef]
- Hiratsuka, J.; Kamitani, N.; Tanaka, R.; Tokiya, R.; Yoden, E.; Sakurai, Y.; Suzuki, M. Long-term outcome of cutaneous melanoma patients treated with boron neutron capture therapy (BNCT). J. Radiat. Res. 2020, 61, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, K.; Okamoto, M.; Kakihana, H.; Mori, Y.; Mishima, Y.; Ichihashi, M.; Tsuji, M.; Nakanishi, T. Study of melanoma seeking agent 10B1-p-boronophenylalanine·HC1 by chemical determination of trace boron in biological materials. KURRI-TR 1985, 260, 233–245. [Google Scholar]
- Tsuboi, T.; Kondoh, H.; Hiratsuka, J.; Mishima, Y. Enhanced melanogenesis induced by tyrosinase gene-transfer increases boron-uptake and killing effect of boron neutron capture therapy for amelanotic melanoma. Pigment Cell Res. 1998, 11, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Shinkai, M.; Honda, H.; Kobayashi, T. Medical application of functionalized magnetic nanoparticles. J. Biosci. Bioeng. 2005, 100, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimer, P.; Balzer, T. Ferucarbotran (Resovist): A new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: Properties, clinical development, and applications. Eur. Radiol. 2003, 13, 1266–1276. [Google Scholar] [CrossRef]
- Castellanos-Rubio, I.; Rodrigo, I.; Olazagoitia-Garmendia, A.; Arriortua, O.; de Muro, I.G.; Garitaonandia, J.S.; Bilbao, J.R.; Fdez-Gubieda, M.L.; Plazaola, F.; Orue, I.; et al. Highly reproducible hyperthermia response in water, agar, and cellular environment by discretely PEGylated magnetite nanoparticles. ACS Appl. Mater. Interfaces. 2020, 12, 27917–27929. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Z.; Jiang, M.; Zhang, C.; Wang, X.; Xiang, L. Light-emitting diode 585 nm photomodulation inhibiting melanin synthesis and inducing autophagy in human melanocytes. J. Dermatol Sci. 2018, 89, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Tamura, Y.; Sato, N.; Yamashita, T.; Takada, T.; Sato, M.; Osai, Y.; Okura, M.; Ono, I.; Ito, A.; et al. Melanoma-targeted chemo-thermo-immuno (CTI)-therapy using N-propionyl-4-S-cysteaminylphenol-magnetite nanoparticles elicits CTL response via heat shock protein-peptide complex release. Cancer Sci. 2010, 101, 1939–1946. [Google Scholar] [CrossRef]
- Takada, T.; Yamashita, T.; Sato, M.; Sato, A.; Ono, I.; Tamura, Y.; Sato, N.; Miyamoto, A.; Ito, A.; Honda, H.; et al. Growth inhibition of re-challenge B16 melanoma transplant by conjugates of melanogenesis substrate and magnetite nanoparticles as the basis for developing melanoma-targeted chemo-thermo-immunotherapy. J. Biomed. Biotechnol. 2009, 2009, 457936. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Shinkai, M.; Honda, H.; Yoshikawa, K.; Saga, S.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Heat shock protein 70 expression induces antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol. Immunother. 2003, 52, 80–88. [Google Scholar] [CrossRef]
- Ito, A.; Shinkai, M.; Hinda, H.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol. Immunother. 2001, 50, 512–522. [Google Scholar] [CrossRef]
- Sato, M.; Yamashita, T.; Ohkura, M.; Osai, Y.; Sato, A.; Takada, T.; Matsusaka, H.; Ono, I.; Tamura, Y.; Sato, N.; et al. N-propionyl-cysteaminylphenol-magnetite conjugate (NPrCAP/M) is a nanoparticle for the targeted growth suppression of melanoma cells. J. Investig. Dermatol. 2009, 129, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef]
- Binder, R.J.; Srivastava, P.K. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 6128–6133. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Yoneda, A.; Takei, N.; Sawada, K. Spatiotemporal regulation of Hsp90-ligand complex leads to immune activation. Front. Immunol. 2016, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- Kato, J.; Uhara, H. Immunotherapy for advanced melanoma: Current situation in Japan. Jpn. J. Clin. Oncol. 2021, 51, 3–9. [Google Scholar] [CrossRef]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; de la Cruz Merino, L.; Khattak, M.A.; Schadendorf, D.; Long, G.V.; et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Basak, E.A.; Vermeer, N.S.; de Joode, K.; Hurkmans, D.P.; Velthuis, D.E.M.; Oomen-de Hoop, E.; Schreurs, M.W.J.; Bins, S.; Koolen, S.L.W.; Debets, R.; et al. Associations between patient and disease characteristics and severe adverse events during immune checkpoint inhibitor treatment: An observational study. Eur. J. Cancer 2022, 174, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.-E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC. Med. 2015, 13, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-term outcomes with Nivolumab plus Ipilimumab or Nivolumab alone versus Ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Jimbow, K.; Szabo, G.; Fitzpatrick, T.B. Ultrastructural investigation of autophagocytosis of melanosomes and programmed death of melanocytes in White Leghorn feathers: A study of morphogenetic events leading to hypomelanosis. Dev. Biol. 1974, 36, 8–23. [Google Scholar] [CrossRef]
- Riley, P.A. Melanogenesis: A realistic target for antimelanoma therapy? Eur. J. Cancer 1991, 27, 1172–1177. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Moy, A.J.; Tunnell, J.W. Combinatorial immunotherapy and nanoparticle mediated hyperthermia. Adv. Drug Deliv. Rev. 2017, 114, 175–183. [Google Scholar] [CrossRef]
- Chao, Y.; Chen, G.; Liang, C.; Xu, J.; Dong, Z.; Han, X.; Wang, C.; Liu, Z. Iron nanoparticles for low-power local magnetic hyperthermia in combination with immune checkpoint blockade for systemic antitumor therapy. Nano Lett. 2019, 19, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. TLR agonists for cancer immunotherapy: Tipping the balance between the immune stimulatory and inhibitory effects. Front. Immunol. 2014, 5, 83. [Google Scholar] [CrossRef] [PubMed]










| Patient no. | Duration of Treatment (hrs) # | Plasma Level (×10−5 M) | Tumor (Percent Labeling Index) | |
|---|---|---|---|---|
| Preinfusion | Postinfusion | |||
| 1 | 120 | 4.0 | 2.0 | 0.2 |
| 2 | 72 | 5.2 | 3.0 | 0.2 |
| 3 | 48 | 3.5 | 1.0 | 0.1 |
| 4 | 48 | 3.1 | 3.0 | 0.2 |
| Chemical Structure (Customary Name) | Abbreviation | MW | 10B Percent | |
|---|---|---|---|---|
| Natural Abundance | 92% 10B Abundance | |||
 | Dopa borate | 263.00 | 0.729 | 3.512 |
| (Sodium Dopa borate) | ||||
 | BPA | 209.01 | 0.917 | 4.423 |
| (p-Borono-phenylalanine) | ||||
 | BPA HCl salt | 245.48 | 0.781 | 3.764 |
 | Dopeba | 181.99 | 1.054 | 5.082 |
| Reference & No. (Years of Study) | Number of Patients | Melanoma Stage | BPA Dose (mg/kg) | Administration Methods | % Tumor Response (Case Responded) |
|---|---|---|---|---|---|
| Fukuda [91] (1987–2002) | 22 | II–IV | 170–210 | iv | CR 68.2% (15/22) PR 23.0% (5/22) |
| Busse [92] (1994–1996) | 4 | III–IV | 400 | oral | CR 25% (1/4) PR 50% (2/4) |
| Menéndez [93] (2003–2007) | 7 | IV | 300 | iv | CR + PR 69.3% (overall Survival: 4 to 23 months) |
| Hiratsuka [94] (2003–2014) | 8 | II | 500 | iv | CR 75% (6/8) PR 25% (2/8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakamatsu, K.; Ito, A.; Tamura, Y.; Hida, T.; Kamiya, T.; Torigoe, T.; Honda, H.; Ito, S.; Jimbow, K. Molecular Events in the Melanogenesis Cascade as Novel Melanoma-Targeted Small Molecules: Principle and Development. Cancers 2022, 14, 5588. https://doi.org/10.3390/cancers14225588
Wakamatsu K, Ito A, Tamura Y, Hida T, Kamiya T, Torigoe T, Honda H, Ito S, Jimbow K. Molecular Events in the Melanogenesis Cascade as Novel Melanoma-Targeted Small Molecules: Principle and Development. Cancers. 2022; 14(22):5588. https://doi.org/10.3390/cancers14225588
Chicago/Turabian StyleWakamatsu, Kazumasa, Akira Ito, Yasuaki Tamura, Tokimasa Hida, Takafumi Kamiya, Toshihiko Torigoe, Hiroyuki Honda, Shosuke Ito, and Kowichi Jimbow. 2022. "Molecular Events in the Melanogenesis Cascade as Novel Melanoma-Targeted Small Molecules: Principle and Development" Cancers 14, no. 22: 5588. https://doi.org/10.3390/cancers14225588
APA StyleWakamatsu, K., Ito, A., Tamura, Y., Hida, T., Kamiya, T., Torigoe, T., Honda, H., Ito, S., & Jimbow, K. (2022). Molecular Events in the Melanogenesis Cascade as Novel Melanoma-Targeted Small Molecules: Principle and Development. Cancers, 14(22), 5588. https://doi.org/10.3390/cancers14225588