
Epigenetics in Pancreatic Ductal Adenocarcinoma: Impact on Biology and Utilization in Diagnostics and Treatment

 , and
, and
Abstract
:Simple Summary
Abstract
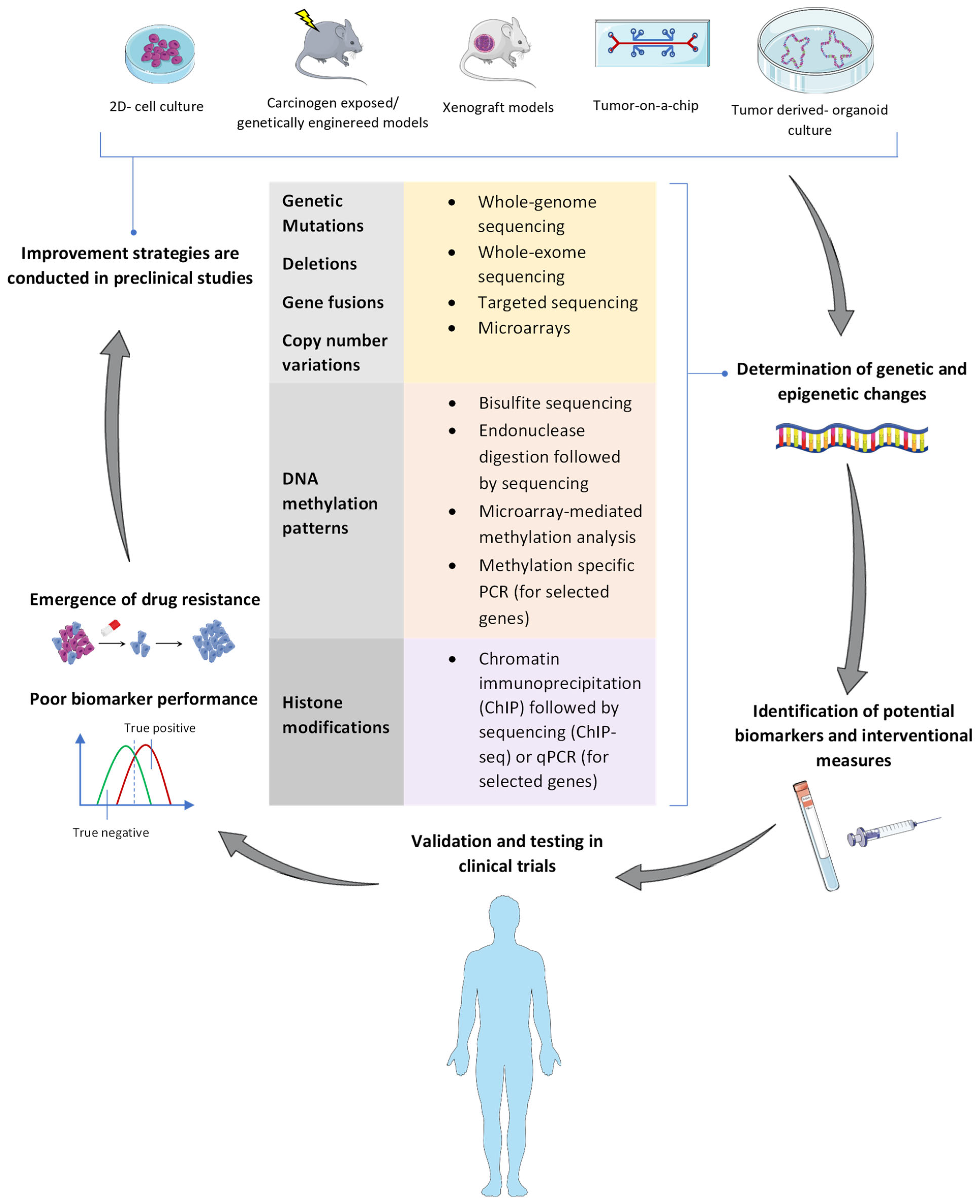
1. Background
2. Epigenetic Modifications in the Pathophysiology of PDAC
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Epigenetic Characteristics of Metastatic PDAC
3. Diagnostic Utility of Epigenetic Modifications in PDAC
3.1. DNA Methylation in Liquid Biopsies as Marker for the Diagnosis of PDAC
3.2. DNA Methylation in Liquid Biopsies as Marker for Prognostication and Treatment Monitoring of PDAC
3.3. Histone Modifications in Liquid Biopsies as Biomarker in PDAC
3.4. Liquid Biopsy to Select Epigenetically Active Treatment in PDAC
4. Epigenetic-Based Therapeutic Approaches
4.1. DNMT Inhibitors (DNMTi)
4.2. HDAC Inhibitors (HDACi)
4.3. Retinoids
4.4. BET Inhibitors (BETi)
4.5. EZH2 Inhibitors (EZH2i)

{kind=link}
| Type | Drug/Route of Administration | Combination | Comparison | Phase (Status) | Condition | Pt number and Results | Reference |
|---|---|---|---|---|---|---|---|
| DNMTi | Decitabine iv | - | - | II (r) | PDAC (unresectable or metastatic) | No results reported | NCT05360264 [105] |
| Decitabine sc | Gemcitabine | - | I (a) | PDAC (metastatic) Sarcoma | No results for PDAC reported | NCT02959164 [113] | |
| Decitabine po | Tetrahydrouridine | - | I (c) | PDAC (metastatic) | 13 pts; 8 evaluable pts: SD n = 1, PD n = 7, median OS 3.1 mo | NCT02847000 [121,122] | |
| Azacitidine sc | Pembrolizumab | - | II (a) | PDAC (unresectable or metastatic) | 36 pts; 34 evaluable pts: PR n = 3, SD n = 8, median OS 4.67 mo 21% ≥ G3 AE | NCT03264404 [117] | |
| Azacitidine sc | Romidepsin nab-Paclitaxel Gemcitabine Durvalumab Lenalidomide | - | I/II (r) | PDAC (metastatic) | No results reported | NCT04257448 [118] | |
| Azacitidine po | - | Observation (OBS) (1:1) | II (c) | PDAC (after adjuvant chemotherapy) | 48 evaluable pts: PFS HR 1.01, OS HR 1.01, median PFS 7.8 mo (AZA) vs. 8.9 mo (OBS), median OS 21.9 mo (AZA) vs. 25.6 mo (OBS) | NCT01845805 [123] | |
| Azacitidine po | Carboplatin nab-Paclitaxel | - | I (c) | Solid tumors | PDAC (part 2): 24 evaluable pts: DCR 46% | NCT01478685 [124] | |
| Guadecitabine iv | Durvalumab | - | I (a) | PDAC HCC BTC | PDAC: 24 evaluable pts: PR n = 1, SD n = 7, median PFS 2.1 mo, median OS 4.4 mo | NCT03257761 [119] | |
| HDACi | Belinostat iv | Carboplatin Paclitaxel | - | I (c) | Solid tumors | PDAC: 3 pts: PR n = 1 | [130] |
| Tacedinaline po | Gemcitabine | Gemcitabine (1:1) | II (c) | PDAC (unresectable or metastatic) | 174 evaluable pts: ORR 12% vs. 14%, OS HR 0.98, median OS 6.5 mo vs. 7.1 mo | NCT00004861 [133] | |
| Vorinostat po | Capecitabine Radiotherapy | - | I (c) | PDAC (resectable, borderline resectable, unresectable) | 21 pts: median OS 13.2 mo | NCT00983268 [139] | |
| Vorinostat po | Marizomib | - | I (c) | PDAC NSCLC Melanoma | PDAC: 2 pts | NCT00667082 [131,140] | |
| Panobinostat po | Bortezomib | - | II (c) | PDAC (metastatic) | 7 evaluable pts: PD n = 7, median PFS 0.86 mo, median OS 4.01 mo | NCT01056601 [134] | |
| Vorinostat po | Bortezomib | - | I (c) | Solid tumors | PDAC: 6 pts | NCT00227513 [135] | |
| Valproic acid po | S-1 | - | I/II (c) | PDAC (unresectable or metastatic) BTC | PDAC: 7 pts | [136] | |
| Mocetinostat po | Gemcitabine | - | I/II (c) | Solid tumors | PDAC: 13 evaluable pts (ph II): SD n = 9, median PFS 5.3 mo, median OS 7.4 mo | NCT00372437 [137] | |
| Resminostat po | S-1 | - | I (c) | PDAC (unresectable or metastatic) BTC | PDAC: 7 pts; 3 evaluable pts (regimen 3): SD n = 2, median PFS 2.3 mo, median OS 4.7 mo | [138] | |
| Vorinostat po | Gemcitabine Sorafenib Radiotherapy | - | I (a) | PDAC (resectable, borderline resectable, unresectable) | 22 pts | NCT02349867 [146] | |
| Romidepsin iv | Gemcitabine | - | I (c) | PDAC (unresectable or metastatic) Other solid tumors | 27 evaluable pts; SD n = 14, PD n = 11; 67% ≥G3 AE | NCT00379639 [132,141] | |
| Romidepsin iv | - | - | I (a) | Solid tumors Lymphoma | PDAC/BTC: 5 pts | NCT01638533 [142] | |
| Entinostat po | Nivolumab | II (c) | PDAC (unresectable or metastatic) BTC | PDAC: 18 evaluable pts: CR/PR n = 3, median OS 3.9 mo; 63% ≥G3 AE | NCT03250273 [145] | ||
| Retinoids | ATRA po | Gemcitabine nab-paclitaxel | - | I (c) | PDAC (unresectable or metastatic) | 28 pts; 15 evaluable pts: PR n = 7, SD n = 7, median OS 11.7 mo; 63% ≥G3 AE | NCT03307148 [162,163] |
| ATRA po | Gemcitabine nab-paclitaxel | Gemcitabine nab-paclitaxel | II (not yet recruiting) | PDAC (unresectable) | No results reported | NCT04241276 [164] | |
| Isotretinoin po | Belinostat | - | I (c) | Solid tumors | PDAC: 3 pts: CR/PR/SD n = 0 | NCT00334789 [165] | |
| ATRA po | Nivolumab | - | I (a) | PDAC (unresectable or metastatic) | No results reported | NCT05482451 [167] | |
| BETi | Mivebresib po | - | - | I (c) | Solid tumors | PDAC: 6 evaluable pts; 56% ≥G3 AE | NCT02391480 [175] |
| Birabresib po | - | - | I (c) | Solid tumors | No results for PDAC reported | NCT02259114 [176] | |
| ZEN-3694 po | Entinostat | - | I/II (r) | Solid tumors Lymphomas | No results reported | NCT05053971 [180] | |
| NUV-868 po | Olaparib Enzalutamide | - | I/II (r) | Solid tumors | No results reported | NCT05252390 [181] | |
| EZH2i | Tazemetostat po | - | - | I (c) | Solid tumors Lymphomas | No results for PDAC reported | NCT01897571 [107] |
| GSK2816126 iv | - | - | I (c) | Solid tumors Lymphomas | PDAC: 2 pts | NCT02082977 [193] | |
| Tazemetostat po | Durvalumab | - | II (r) | Solid tumors | No results reported | NCT04705818 [199] |
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Schouten, T.J.; Daamen, L.A.; Dorland, G.; van Roessel, S.R.; Groot, V.P.; Besselink, M.G.; Bonsing, B.A.; Bosscha, K.; Brosens, L.A.A.; Busch, O.R.; et al. Nationwide Validation of the 8th American Joint Committee on Cancer TNM Staging System and Five Proposed Modifications for Resected Pancreatic Cancer. Ann. Surg. Oncol. 2022, 29, 5988–5999. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; Aprile, G.; Conca, R.; Petrioli, R.; Ramello, M.; Roviello, G. The Impact of Age, Performance Status and Comorbidities on Nab-Paclitaxel plus Gemcitabine Effectiveness in Patients with Metastatic Pancreatic Cancer. Sci. Rep. 2022, 12, 8244. [Google Scholar] [CrossRef] [PubMed]
- Balaban, E.P.; Mangu, P.B.; Yee, N.S. Locally Advanced Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Summary. J. Oncol. Pract. 2017, 13, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; O’Reilly, E.M. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020, 80, 647–669. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. P16 and K-Ras Gene Mutations in the Intraductal Precursors of Human Pancreatic Adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar]
- DiGiuseppe, J.A.; Redston, M.S.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. P53-Independent Expression of the Cyclin-Dependent Kinase Inhibitor P21 in Pancreatic Carcinoma. Am. J. Pathol. 1995, 147, 884–888. [Google Scholar]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Sina-Frey, M.; Lang, S.; Wild, A.; Gerdes, B.; Barth, P.; Kress, R.; Grützmann, R.; Colombo-Benkmann, M.; Ziegler, A.; et al. CDKN2A Germline Mutations in Familial Pancreatic Cancer. Ann. Surg. 2002, 236, 730–737. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct Epigenetic Landscapes Underlie the Pathobiology of Pancreatic Cancer Subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLoS ONE 2015, 10, e0128814. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited Heterogeneity of Known Driver Gene Mutations among the Metastases of Individual Patients with Pancreatic Cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Embuscado, E.E.; Laheru, D.; Ricci, F.; Yun, K.J.; de Boom Witzel, S.; Seigel, A.; Flickinger, K.; Hidalgo, M.; Bova, G.S.; Iacobuzio-Donahue, C.A. Immortalizing the Complexity of Cancer Metastasis Genetic Features of Lethal Metastatic Pancreatic Cancer Obtained from Rapid Autopsy. Cancer Biol. Ther. 2005, 4, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Waddington, C.H. The Epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Licht, J.D.; Bennett, R.L. Leveraging Epigenetics to Enhance the Efficacy of Immunotherapy. Clin. Epigenetics 2021, 13, 115. [Google Scholar] [CrossRef]
- Wang, S.S.; Xu, J.; Ji, K.Y.; Hwang, C.-I. Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules 2021, 11, 1082. [Google Scholar] [CrossRef]
- Hayashi, A.; Fan, J.; Chen, R.; Ho, Y.; Makohon-Moore, A.P.; Lecomte, N.; Zhong, Y.; Hong, J.; Huang, J.; Sakamoto, H.; et al. A Unifying Paradigm for Transcriptional Heterogeneity and Squamous Features in Pancreatic Ductal Adenocarcinoma. Nat. Cancer 2020, 1, 59–74. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Large-Scale Epigenomic Reprogramming during Pancreatic Cancer Progression Links Anabolic Glucose Metabolism to Distant Metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Fundamental Role of Epigenetic Events in Cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA Methylation of Intragenic CpG Islands Depends on Their Transcriptional Activity during Differentiation and Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A.; Gegonne, A.; Jones, P.L.; Ballestar, E.; Aubry, F.; Wolffe, A.P. Mi-2 Complex Couples DNA Methylation to Chromatin Remodelling and Histone Deacetylation. Nat. Genet. 1999, 23, 62–66. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for de Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET Proteins and the Control of Cytosine Demethylation in Cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.C.; Jimeno, A.; Lin, S.H.; Wheelhouse, J.; Chan, F.; Solomon, A.; Rajeshkumar, N.V.; Rubio-Viqueira, B.; Hidalgo, M. Characterizing DNA Methylation Patterns in Pancreatic Cancer Genome. Mol. Oncol. 2009, 3, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, Y.; Kisiel, J.B.; Mahoney, D.W.; Michaud, D.S.; Guo, X.; Taylor, W.R.; Shu, X.-O.; Shu, X.; Liu, D.; et al. Integrating Genome and Methylome Data to Identify Candidate DNA Methylation Biomarkers for Pancreatic Cancer Risk. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 2079–2087. [Google Scholar] [CrossRef]
- Ozturk, H.; Cingoz, H.; Tufan, T.; Yang, J.; Adair, S.J.; Tummala, K.S.; Kuscu, C.; Kinali, M.; Comertpay, G.; Nagdas, S.; et al. ISL2 Is a Putative Tumor Suppressor Whose Epigenetic Silencing Reprograms the Metabolism of Pancreatic Cancer. Dev. Cell 2022, 57, 1331–1346.e9. [Google Scholar] [CrossRef]
- Sato, N.; Maitra, A.; Fukushima, N.; van Heek, N.T.; Matsubayashi, H.; Iacobuzio-Donahue, C.A.; Rosty, C.; Goggins, M. Frequent Hypomethylation of Multiple Genes Overexpressed in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2003, 63, 4158–4166. [Google Scholar] [PubMed]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Büscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2021, 11, 638–659. [Google Scholar] [CrossRef] [PubMed]
- Eyres, M.; Lanfredini, S.; Xu, H.; Burns, A.; Blake, A.; Willenbrock, F.; Goldin, R.; Hughes, D.; Hughes, S.; Thapa, A.; et al. TET2 Drives 5hmc Marking of GATA6 and Epigenetically Defines Pancreatic Ductal Adenocarcinoma Transcriptional Subtypes. Gastroenterology 2021, 161, 653–668.e16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Huang, C.; Ma, T.-T.; Bian, E.-B.; He, Y.; Zhang, L.; Li, J. SOCS1 Hypermethylation Mediated by DNMT1 Is Associated with Lipopolysaccharide-Induced Inflammatory Cytokines in Macrophages. Toxicol. Lett. 2014, 225, 488–497. [Google Scholar] [CrossRef]
- Tang, R.-Z.; Zhu, J.-J.; Yang, F.-F.; Zhang, Y.-P.; Xie, S.-A.; Liu, Y.-F.; Yao, W.-J.; Pang, W.; Han, L.-L.; Kong, W.; et al. DNA Methyltransferase 1 and Krüppel-like Factor 4 Axis Regulates Macrophage Inflammation and Atherosclerosis. J. Mol. Cell Cardiol. 2019, 128, 11–24. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, X.; Fujiwara, K.; Jurcak, N.; Muth, S.; Zhou, J.; Xiao, Q.; Li, A.; Che, X.; Li, Z.; et al. Pancreatic Cancer Cells Render Tumor-Associated Macrophages Metabolically Reprogrammed by a GARP and DNA Methylation-Mediated Mechanism. Signal Transduct. Target Ther. 2021, 6, 366. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Hazelton, W.D.; Luebeck, G.E.; Grady, W.M. Epigenetic Aging: More Than Just a Clock When It Comes to Cancer. Cancer Res. 2020, 80, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Cancer of the Pancreas—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 13 November 2022).
- Raffenne, J.; Martin, F.A.; Nicolle, R.; Konta, M.; Blum, Y.; Torrisani, J.; Puleo, F.; Bachet, J.B.; Svrcek, M.; Bardier-Dupas, A.; et al. Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features. Cancers 2021, 13, 1234. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Ruan, M.; Zhao, N.; Koestler, D.C.; De Vivo, I.; Kelsey, K.T.; Michaud, D.S. DNA Methylation Ageing Clocks and Pancreatic Cancer Risk: Pooled Analysis of Three Prospective Nested Case-Control Studies. Epigenetics 2021, 16, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic Modifications of Histones in Cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [Green Version]
- Köenig, A.; Linhart, T.; Schlengemann, K.; Reutlinger, K.; Wegele, J.; Adler, G.; Singh, G.; Hofmann, L.; Kunsch, S.; Büch, T.; et al. NFAT-Induced Histone Acetylation Relay Switch Promotes c-Myc-Dependent Growth in Pancreatic Cancer Cells. Gastroenterology 2010, 138, 1189–1199.e2. [Google Scholar] [CrossRef] [Green Version]
- Mees, S.T.; Mardin, W.A.; Wendel, C.; Baeumer, N.; Willscher, E.; Senninger, N.; Schleicher, C.; Colombo-Benkmann, M.; Haier, J. EP300--a MiRNA-Regulated Metastasis Suppressor Gene in Ductal Adenocarcinomas of the Pancreas. Int. J. Cancer 2010, 126, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.-H.; Xu, X.-G.; Yan, S.-L.; Sun, Z.; Ying, Y.; Wang, B.-K.; Tu, Y.-X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Role of Histone Deacetylases in Pancreas: Implications for Pathogenesis and Therapy. World J. Gastrointest. Oncol. 2015, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Measures, A.R.; Measures, A.M.; Wilson, B.G.; Cortopassi, W.A.; Alexander, R.; Höss, M.; Hewings, D.S.; Rooney, T.P.C.; Paton, R.S.; et al. Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. ACS Chem. Biol. 2015, 10, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Yan, J.; Diaz, J.; Jiao, J.; Wang, R.; You, J. Perturbation of BRD4 Protein Function by BRD4-NUT Protein Abrogates Cellular Differentiation in NUT Midline Carcinoma. J. Biol. Chem. 2011, 286, 27663–27675. [Google Scholar] [CrossRef] [Green Version]
- Junwei, S.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The Role of Histone Methylation in the Development of Digestive Cancers: A Potential Direction for Cancer Management. Signal Transduct. Target Ther. 2020, 5, 143. [Google Scholar] [CrossRef] [PubMed]
- Benitz, S.; Straub, T.; Mahajan, U.M.; Mutter, J.; Czemmel, S.; Unruh, T.; Wingerath, B.; Deubler, S.; Fahr, L.; Cheng, T.; et al. Ring1b-Dependent Epigenetic Remodelling Is an Essential Prerequisite for Pancreatic Carcinogenesis. Gut 2019, 68, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.A.; Dhele, N.; Cheemadan, S.; Ketkar, A.; Jayandharan, G.R.; Palakodeti, D.; Rampalli, S. Ezh2 Mediated H3K27me3 Activity Facilitates Somatic Transition during Human Pluripotent Reprogramming. Sci. Rep. 2015, 5, 8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of Pancreatic Tumor Cell Proliferation and Chemoresistance by the Histone Methyltransferase EZH2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Küffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. EZH2 Regulates Pancreatic Cancer Subtype Identity and Tumor Progression via Transcriptional Repression of GATA6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical Significance of the Genetic Landscape of Pancreatic Cancer and Implications for Identification of Potential Long Term Survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [Green Version]
- Miquel, M.; Zhang, S.; Pilarsky, C. Pre-Clinical Models of Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 748631. [Google Scholar] [CrossRef] [PubMed]
- Toll, A.D.; Dasgupta, A.; Potoczek, M.; Yeo, C.J.; Kleer, C.G.; Brody, J.R.; Witkiewicz, A.K. Implications of Enhancer of Zeste Homologue 2 Expression in Pancreatic Ductal Adenocarcinoma. Hum. Pathol. 2010, 41, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Jiao, F.; Hu, H.; Yuan, C.; Wang, L.; Jin, Z.-L.; Song, W.; Wang, L.-W. EZH2 Promotes Cell Migration and Invasion but Not Alters Cell Proliferation by Suppressing E-Cadherin, Partly through Association with MALAT-1 in Pancreatic Cancer. Oncotarget 2016, 7, 11194–11207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-Cadherin Regulates Metastasis of Pancreatic Cancer in Vivo and Is Suppressed by a SNAIL/HDAC1/HDAC2 Repressor Complex. Gastroenterology 2009, 137, 361–371. [Google Scholar] [CrossRef]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.; Heidecke, C.-D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of Histone Deacetylases HDAC1 and HDAC2 by the Transcriptional Repressor ZEB1 Downregulates E-Cadherin Expression in Pancreatic Cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Washington, M.K.; Crawford, H.C. Loss of FOXA1/2 Is Essential for the Epithelial-to-Mesenchymal Transition in Pancreatic Cancer. Cancer Res. 2010, 70, 2115–2125. [Google Scholar] [CrossRef] [Green Version]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic Inactivation of TFPI-2 as a Common Mechanism Associated with Growth and Invasion of Pancreatic Ductal Adenocarcinoma. Oncogene 2005, 24, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Fukushima, N.; Chang, R.; Matsubayashi, H.; Goggins, M. Differential and Epigenetic Gene Expression Profiling Identifies Frequent Disruption of the RELN Pathway in Pancreatic Cancers. Gastroenterology 2006, 130, 548–565. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Song, S.; Patch, A.-M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-Wide DNA Methylation Patterns in Pancreatic Ductal Adenocarcinoma Reveal Epigenetic Deregulation of SLIT-ROBO, ITGA2 and MET Signaling. Int. J. Cancer 2014, 135, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-M.; Wang, J.-S.; Zulfiqar, H.; Lv, H.; Dao, F.-Y.; Lin, H. Early Diagnosis of Pancreatic Ductal Adenocarcinoma by Combining Relative Expression Orderings with Machine-Learning Method. Front. Cell Dev. Biol. 2020, 8, 582864. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic Ductal Adenocarcinoma: Biological Hallmarks, Current Status, and Future Perspectives of Combined Modality Treatment Approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in Diagnosis of Pancreatic Cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- George, B.; Haberberger, J.; Ferguson, N.L.; Gjoerup, O.; McGregor, K.; Hendifar, A.E.; Laheru, D.A.; Weekes, C.D.; Ross, J.S.; Hemmerich, A. Correlation between Comprehensive Genomic Profiling (CGP) Utilizing Tissue-Based Testing (T-CGP) and Cell-Free DNA (CfDNA) in Patients (Pts) with Pancreatic Ductal Adenocarcinoma (PDAC). J. Clin. Oncol. 2021, 39 (Suppl. 3), 422. [Google Scholar] [CrossRef]
- Hipp, J.; Hussung, S.; Timme-Bronsert, S.; Boerries, M.; Biesel, E.; Fichtner-Feigl, S.; Fritsch, R.; Wittel, U.A. Perioperative Cell-Free Mutant KRAS Dynamics in Patients with Pancreatic Cancer. Br. J. Surg. 2021, 108, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Hussung, S.; Akhoundova, D.; Hipp, J.; Follo, M.; Klar, R.F.U.; Philipp, U.; Scherer, F.; von Bubnoff, N.; Duyster, J.; Boerries, M.; et al. Longitudinal Analysis of Cell-Free Mutated KRAS and CA 19-9 Predicts Survival Following Curative Resection of Pancreatic Cancer. BMC Cancer 2021, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, J.; Philipp, U.; Jolic, M.; Follo, M.; Hussung, S.; Waldeck, S.; Deuter, M.; Rassner, M.; Braune, J.; Rawluk, J.; et al. Personalized Treatment Selection and Disease Monitoring Using Circulating Tumor DNA Profiling in Real-World Cancer Patient Management. Diagnostics 2020, 10, 550. [Google Scholar] [CrossRef]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic Cancer: Circulating Tumor Cells and Primary Tumors Show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, A.A.; Scholtens, D.; Talamonti, M.S.; Bentrem, D.J.; Levenson, V.V. Methylation Profile of Circulating Plasma DNA in Patients with Pancreatic Cancer. J. Surg. Oncol. 2009, 99, 119–122. [Google Scholar] [CrossRef]
- Park, J.W.; Baek, I.H.; Kim, Y.T. Preliminary Study Analyzing the Methylated Genes in the Plasma of Patients with Pancreatic Cancer. Scand. J. Surg. 2012, 101, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.K.; Ryu, J.K.; Yoon, W.J.; Lee, S.H.; Lee, G.Y.; Jeong, K.-S.; Kim, Y.-T.; Yoon, Y.B. The Role of Quantitative NPTX2 Hypermethylation as a Novel Serum Diagnostic Marker in Pancreatic Cancer. Pancreas 2012, 41, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Rashid, S.; Rashid, S.; Dash, N.R.; Gupta, S.; Saraya, A. Clinical Significance of Promoter Methylation Status of Tumor Suppressor Genes in Circulating DNA of Pancreatic Cancer Patients. J. Cancer Res. Clin. Oncol. 2020, 146, 897–907. [Google Scholar] [CrossRef]
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter Methylation of ADAMTS1 and BNC1 as Potential Biomarkers for Early Detection of Pancreatic Cancer in Blood. Clin. Epigenetics 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel Methylation Biomarker Panel for the Early Detection of Pancreatic Cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Drewes, A.M.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-Free DNA Promoter Hypermethylation in Plasma as a Diagnostic Marker for Pancreatic Adenocarcinoma. Clin. Epigenetics 2016, 8, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of Tissue-Specific Cell Death Using Methylation Patterns of Circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Xu, J.; Link, A.; Matsuyama, T.; Yamamura, K.; Parker, M.I.; Uetake, H.; Balaguer, F.; Borazanci, E.; Tsai, S.; et al. EpiPanGI Dx: A Cell-Free DNA Methylation Fingerprint for the Early Detection of Gastrointestinal Cancers. Clin. Cancer Res. 2021, 27, 6135–6144. [Google Scholar] [CrossRef]
- Majumder, S.; Taylor, W.R.; Foote, P.H.; Berger, C.K.; Wu, C.W.; Mahoney, D.W.; Bamlet, W.R.; Burger, K.N.; Postier, N.; de la Fuente, J.; et al. High Detection Rates of Pancreatic Cancer Across Stages by Plasma Assay of Novel Methylated DNA Markers and CA19-9. Clin. Cancer Res. 2021, 27, 2523–2532. [Google Scholar] [CrossRef]
- Vrba, L.; Futscher, B.W.; Oshiro, M.; Watts, G.S.; Menashi, E.; Hu, C.; Hammad, H.; Pennington, D.R.; Golconda, U.; Gavini, H.; et al. Liquid Biopsy, Using a Novel DNA Methylation Signature, Distinguishes Pancreatic Adenocarcinoma from Benign Pancreatic Disease. Clin. Epigenetics 2022, 14, 28. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Suehiro, Y.; Kaino, S.; Suenaga, S.; Tsuyama, T.; Matsui, H.; Higaki, S.; Fujii, I.; Suzuki, C.; Hoshida, T.; et al. Combination of CA19-9 and Blood Free-Circulating Methylated RUNX3 May Be Useful to Diagnose Stage I Pancreatic Cancer. Oncology 2021, 99, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, S.D.; Thorlacius-Ussing, O. Cell-Free DNA Methylation as Blood-Based Biomarkers for Pancreatic Adenocarcinoma-A Literature Update. Epigenomes 2021, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Raimondo, M.; Taylor, W.R.; Yab, T.C.; Berger, C.K.; Dukek, B.A.; Cao, X.; Foote, P.H.; Wu, C.W.; Devens, M.E.; et al. Methylated DNA in Pancreatic Juice Distinguishes Patients with Pancreatic Cancer from Controls. Clin. Gastroenterol. Hepatol. 2020, 18, 676–683.e3. [Google Scholar] [CrossRef]
- Majumder, S.; Taylor, W.R.; Yab, T.C.; Berger, C.K.; Dukek, B.A.; Cao, X.; Foote, P.H.; Wu, C.W.; Mahoney, D.W.; Aslanian, H.R.; et al. Novel Methylated DNA Markers Discriminate Advanced Neoplasia in Pancreatic Cysts: Marker Discovery, Tissue Validation, and Cyst Fluid Testing. Am. J. Gastroenterol. 2019, 114, 1539–1549. [Google Scholar] [CrossRef]
- Kisiel, J.B.; Yab, T.C.; Taylor, W.R.; Chari, S.T.; Petersen, G.M.; Mahoney, D.W.; Ahlquist, D.A. Stool DNA Testing for the Detection of Pancreatic Cancer: Assessment of Methylation Marker Candidates. Cancer 2012, 118, 2623–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Promoter Hypermethylation in Plasma-Derived Cell-Free DNA as a Prognostic Marker for Pancreatic Adenocarcinoma Staging. Int. J. Cancer 2017, 141, 2489–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Johansen, M.B.; Pedersen, I.S.; Krarup, H.; Thorlacius-Ussing, O. Cell-Free DNA Promoter Hypermethylation in Plasma as a Predictive Marker for Survival of Patients with Pancreatic Adenocarcinoma. Oncotarget 2017, 8, 93942–93956. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhu, R.; Sun, W.; Wang, J.; Liu, J. Analysis of Methylation-driven Genes in Pancreatic Ductal Adenocarcinoma for Predicting Prognosis. J. Cancer 2021, 12, 6507–6518. [Google Scholar] [CrossRef]
- Pietrasz, D.; Wang-Renault, S.; Taieb, J.; Dahan, L.; Postel, M.; Durand-Labrunie, J.; Le Malicot, K.; Mulot, C.; Rinaldi, Y.; Phelip, J.-M.; et al. Prognostic Value of Circulating Tumour DNA in Metastatic Pancreatic Cancer Patients: Post-Hoc Analyses of Two Clinical Trials. Br. J. Cancer 2022, 126, 440–448. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Holdenrieder, S.; Stieber, P. Clinical Use of Circulating Nucleosomes. Crit. Rev. Clin. Lab. Sci. 2009, 46, 1–24. [Google Scholar] [CrossRef]
- Bauden, M.; Pamart, D.; Ansari, D.; Herzog, M.; Eccleston, M.; Micallef, J.; Andersson, B.; Andersson, R. Circulating Nucleosomes as Epigenetic Biomarkers in Pancreatic Cancer. Clin. Epigenetics 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Mottini, C.; Tomihara, H.; Carrella, D.; Lamolinara, A.; Iezzi, M.; Huang, J.K.; Amoreo, C.A.; Buglioni, S.; Manni, I.; Robinson, F.S.; et al. Predictive Signatures Inform the Effective Repurposing of Decitabine to Treat KRAS-Dependent Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 5612–5625. [Google Scholar] [CrossRef] [Green Version]
- A Proof-of-Concept, Biomarker-Driven, Phase-II Clinical Trial to Explore the Activity of Decitabine Repurposing Against Advanced, Refractory, KRAS-Dependent Pancreatic Ductal Adenocarcinoma (PDAC): The ORIENTATE Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT05360264 (accessed on 17 August 2022).
- Tsuda, M.; Fukuda, A.; Kawai, M.; Araki, O.; Seno, H. The Role of the SWI/SNF Chromatin Remodeling Complex in Pancreatic Ductal Adenocarcinoma. Cancer Sci. 2021, 112, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma and Advanced Solid Tumours: A First-in-Human, Open-Label, Phase 1 Study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Armstrong, K.; Drew, A.; Grassian, A.R.; Feldman, I.; Knutson, S.K.; Kuplast-Barr, K.; Roche, M.; Campbell, J.; Ho, P.; et al. Selective Killing of SMARCA2- and SMARCA4-Deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2: In Vitro and In Vivo Preclinical Models. Mol. Cancer Ther. 2017, 16, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating Agents (HMA) for the Treatment of Acute Myeloid Leukemia and Myelodysplastic Syndromes: Mechanisms of Resistance and Novel HMA-Based Therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Becker, H.; Pfeifer, D.; Ihorst, G.; Pantic, M.; Wehrle, J.; Rüter, B.H.; Bullinger, L.; Hackanson, B.; Germing, U.; Kuendgen, A.; et al. Monosomal Karyotype and Chromosome 17p Loss or TP53 Mutations in Decitabine-Treated Patients with Acute Myeloid Leukemia. Ann. Hematol. 2020, 99, 1551–1560. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Hatada, I.; Nakagama, H.; Ochiya, T. Epigenetic Reprogramming Using 5-Azacytidine Promotes an Anti-Cancer Response in Pancreatic Adenocarcinoma Cells. Cell Death Dis. 2018, 9, 468. [Google Scholar] [CrossRef] [Green Version]
- A Phase 1b Study: Treatment of Refractory Pancreatic Adenocarcinoma and Advanced Soft Tissue or Bone Sarcomas Using Decitabine Combined with Gemcitabine. Available online: https://clinicaltrials.gov/ct2/show/NCT02959164 (accessed on 17 August 2022).
- Shakya, R.; Gonda, T.; Quante, M.; Salas, M.; Kim, S.; Brooks, J.; Hirsch, S.; Davies, J.; Cullo, A.; Olive, K.; et al. Hypomethylating Therapy in an Aggressive Stroma-Rich Model of Pancreatic Carcinoma. Cancer Res. 2013, 73, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonda, T.A.; Fang, J.; Salas, M.; Do, C.; Hsu, E.; Zhukovskaya, A.; Siegel, A.; Takahashi, R.; Lopez-Bujanda, Z.A.; Drake, C.G.; et al. A DNA Hypomethylating Drug Alters the Tumor Microenvironment and Improves the Effectiveness of Immune Checkpoint Inhibitors in a Mouse Model of Pancreatic Cancer. Cancer Res. 2020, 80, 4754–4767. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, N.D.; Zuniga, E.; Johnson, B.L.; Diamond, D.J.; Manuel, E.R. 5-Azacytidine Potentiates Anti-Tumor Immunity in a Model of Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Safyan, R.A.; Manji, G.A.; Lee, S.M.; Silva, R.; Bates, S.E.; White, R.A.; Jamison, J.K.R.; Bass, A.J.; Schwartz, G.K.; Oberstein, P.E.; et al. Phase 2 Study of Azacitidine (AZA) plus Pembrolizumab (Pembro) as Second-Line Treatment in Patients with Advanced Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2022, 40 (Suppl. 16), 4158. [Google Scholar] [CrossRef]
- A Multicenter, Phase I/II Study of Sequential Epigenetic and Immune Targeting in Combination with Nab-Paclitaxel/Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04257448 (accessed on 17 August 2022).
- Algaze, S.; Hanna, D.L.; Azad, N.S.; Thomas, J.S.; Iqbal, S.; Habib, D.; Ning, Y.; Barzi, A.; Patel, R.; Lenz, H.-J.; et al. A Phase Ib Study of Guadecitabine and Durvalumab in Patients with Advanced Hepatocellular Carcinoma, Pancreatic Adenocarcinoma, and Biliary Cancers. J. Clin. Oncol. 2022, 40 (Suppl. 4), 574. [Google Scholar] [CrossRef]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA Expression/Activity in Males Contributes to Decreased Cytidine Analogue Half-Life and Likely Contributes to Worse Outcomes with 5-Azacytidine or Decitabine Therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Sohal, D.; Krishnamurthi, S.; Tohme, R.; Gu, X.; Lindner, D.; Landowski, T.H.; Pink, J.; Radivoyevitch, T.; Fada, S.; Lee, Z.; et al. A Pilot Clinical Trial of the Cytidine Deaminase Inhibitor Tetrahydrouridine Combined with Decitabine to Target DNMT1 in Advanced, Chemorefractory Pancreatic Cancer. Am. J. Cancer Res. 2020, 10, 3047–3060. [Google Scholar]
- P53/P16-Independent Epigenetic Therapy with Oral Decitabine/Tetrahydrouridine for Advanced Pancreatic Cancer That Has Progressed through One or More Lines of Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02847000 (accessed on 17 August 2022).
- Heumann, T.R.; Baretti, M.; Sugar, E.; Durhman, J.; Liden, S.; Miles, T.; Lopez-Vidal, T.Y.; Leatherman, J.; Sharma, A.; Ahuja, N.; et al. 1470P Oral Azacitidine (CC-486) in Patients with Resected Pancreatic Adenocarcinoma at High Risk for Recurrence. Ann. Oncol. 2021, 32, S1087–S1088. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Rasco, D.W.; Heath, E.I.; Munster, P.N.; Schellens, J.H.M.; Isambert, N.; Le Tourneau, C.; O’Neil, B.; Mathijssen, R.H.J.; Lopez-Martin, J.A.; et al. Phase I Study of CC-486 Alone and in Combination with Carboplatin or Nab-Paclitaxel in Patients with Relapsed or Refractory Solid Tumors. Clin. Cancer Res. 2018, 24, 4072–4080. [Google Scholar] [CrossRef] [Green Version]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Garmpi, A.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; et al. Histone Deacetylase (HDAC) Inhibitors: Current Evidence for Therapeutic Activities in Pancreatic Cancer. Anticancer Res. 2015, 35, 3129–3135. [Google Scholar]
- García-Morales, P.; Gómez-Martínez, A.; Carrato, A.; Martínez-Lacaci, I.; Barberá, V.M.; Soto, J.L.; Carrasco-García, E.; Menéndez-Gutierrez, M.P.; Castro-Galache, M.D.; Ferragut, J.A.; et al. Histone Deacetylase Inhibitors Induced Caspase-Independent Apoptosis in Human Pancreatic Adenocarcinoma Cell Lines. Mol. Cancer Ther. 2005, 4, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffon, C. Histone Deacetylase Inhibition Restores Expression of Hypoxia-Inducible Protein NDRG1 in Pancreatic Cancer. Pancreas 2018, 47, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maietta, I.; Martínez-Pérez, A.; Álvarez, R.; De Lera, Á.R.; González-Fernández, Á.; Simón-Vázquez, R. Synergistic Antitumoral Effect of Epigenetic Inhibitors and Gemcitabine in Pancreatic Cancer Cells. Pharmaceuticals 2022, 15, 824. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Demirjian, A.; Sui, J.; Marasco, W.; Callery, M.P. Histone Deacetylase Inhibitor Trichostatin A and Proteasome Inhibitor PS-341 Synergistically Induce Apoptosis in Pancreatic Cancer Cells. Biochem. Biophys. Res. Commun. 2006, 348, 1245–1253. [Google Scholar] [CrossRef]
- Lassen, U.; Molife, L.R.; Sorensen, M.; Engelholm, S.-A.; Vidal, L.; Sinha, R.; Penson, R.T.; Buhl-Jensen, P.; Crowley, E.; Tjornelund, J.; et al. A Phase I Study of the Safety and Pharmacokinetics of the Histone Deacetylase Inhibitor Belinostat Administered in Combination with Carboplatin and/or Paclitaxel in Patients with Solid Tumours. Br. J. Cancer 2010, 103, 12–17. [Google Scholar] [CrossRef]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 Clinical Trial of the Novel Proteasome Inhibitor Marizomib with the Histone Deacetylase Inhibitor Vorinostat in Patients with Melanoma, Pancreatic and Lung Cancer Based on in Vitro Assessments of the Combination. Invest. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Jones, S.F.; Infante, J.R.; Spigel, D.R.; Peacock, N.W.; Thompson, D.S.; Greco, F.A.; McCulloch, W.; Burris, H.A. Phase 1 Results from a Study of Romidepsin in Combination with Gemcitabine in Patients with Advanced Solid Tumors. Cancer Invest. 2012, 30, 481–486. [Google Scholar] [CrossRef]
- Richards, D.A.; Boehm, K.A.; Waterhouse, D.M.; Wagener, D.J.; Krishnamurthi, S.S.; Rosemurgy, A.; Grove, W.; Macdonald, K.; Gulyas, S.; Clark, M.; et al. Gemcitabine plus CI-994 Offers No Advantage over Gemcitabine Alone in the Treatment of Patients with Advanced Pancreatic Cancer: Results of a Phase II Randomized, Double-Blind, Placebo-Controlled, Multicenter Study. Ann. Oncol. 2006, 17, 1096–1102. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II Study of Panobinostat and Bortezomib in Patients with Pancreatic Cancer Progressing on Gemcitabine-Based Therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar]
- Deming, D.A.; Ninan, J.; Bailey, H.H.; Kolesar, J.M.; Eickhoff, J.; Reid, J.M.; Ames, M.M.; McGovern, R.M.; Alberti, D.; Marnocha, R.; et al. A Phase I Study of Intermittently Dosed Vorinostat in Combination with Bortezomib in Patients with Advanced Solid Tumors. Invest. New Drugs 2014, 32, 323–329. [Google Scholar] [CrossRef]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of Valproic Acid in Combination with S-1 on Advanced Pancreatobiliary Tract Cancers: Clinical Study Phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II Study of Mocetinostat in Combination with Gemcitabine for Patients with Advanced Pancreatic Cancer and Other Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ohno, I.; Ueno, H.; Mitsunaga, S.; Hashimoto, Y.; Okusaka, T.; Kondo, S.; Sasaki, M.; Sakamoto, Y.; Takahashi, H.; et al. Phase I Study of Resminostat, an HDAC Inhibitor, Combined with S-1 in Patients with Pre-Treated Biliary Tract or Pancreatic Cancer. Invest. New Drugs 2019, 37, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I Trial of Vorinostat Added to Chemoradiation with Capecitabine in Pancreatic Cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NPI-0052 and Vorinostat in Patients with Non-Small Cell Lung Cancer, Pancreatic Cancer, Melanoma or Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00667082 (accessed on 17 August 2022).
- A Phase I/II Study of Romidepsin (Depsipeptide) in Combination with Gemcitabine in Patients with Pancreatic and Other Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT00379639 (accessed on 17 August 2022).
- Connolly, R.M.; Laille, E.; Vaishampayan, U.; Chung, V.; Kelly, K.; Dowlati, A.; Alese, O.B.; Harvey, R.D.; Haluska, P.; Siu, L.L.; et al. Phase I and Pharmacokinetic Study of Romidepsin in Patients with Cancer and Hepatic Dysfunction: A National Cancer Institute Organ Dysfunction Working Group Study. Clin. Cancer Res. 2020, 26, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic Enhancement of Antigen Processing and Presentation Promotes Immune Recognition of Tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [Green Version]
- A Phase 2 Clinical Trial of Entinostat in Combination with Nivolumab for Patients with Previously Treated Unresectable or Metastatic Cholangiocarcinoma and Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03250273 (accessed on 17 August 2022).
- Poklepovic, A.S.; Fields, E.C.; Bandyopadhyay, D.; Tombes, M.B.; Kmieciak, M.; McGuire, W.P.; Gordon, S.W.; Kaplan, B.J.; Myers, J.L.; Matin, K.; et al. A Phase 1 Study of Neoadjuvant Chemotherapy Followed by Concurrent Chemoradiation with Gemcitabine, Sorafenib, and Vorinostat in Pancreatic Cancer. J. Clin. Oncol. 2021, 39, e16268. [Google Scholar] [CrossRef]
- Streubel, G.; Schrepfer, S.; Kallus, H.; Parnitzke, U.; Wulff, T.; Hermann, F.; Borgmann, M.; Hamm, S. Histone Deacetylase Inhibitor Resminostat in Combination with Sorafenib Counteracts Platelet-Mediated pro-Tumoral Effects in Hepatocellular Carcinoma. Sci. Rep. 2021, 11, 9587. [Google Scholar] [CrossRef]
- Ischenko, I.; Petrenko, O.; Hayman, M.J. A MEK/PI3K/HDAC Inhibitor Combination Therapy for KRAS Mutant Pancreatic Cancer Cells. Oncotarget 2015, 6, 15814–15827. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Hausmann, S.; Lyu, R.; Li, T.-M.; Lofgren, S.M.; Flores, N.M.; Fuentes, M.E.; Caporicci, M.; Yang, Z.; Meiners, M.J.; et al. SETD5-Coordinated Chromatin Reprogramming Regulates Adaptive Resistance to Targeted Pancreatic Cancer Therapy. Cancer Cell 2020, 37, 834–849.e13. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of Acute Promyelocytic Leukemia: Updated Recommendations from an Expert Panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lübbert, M.; Grishina, O.; Schmoor, C.; Schlenk, R.F.; Jost, E.; Crysandt, M.; Heuser, M.; Thol, F.; Salih, H.R.; Schittenhelm, M.M.; et al. Valproate and Retinoic Acid in Combination with Decitabine in Elderly Nonfit Patients with Acute Myeloid Leukemia: Results of a Multicenter, Randomized, 2 × 2, Phase II Trial. J. Clin. Oncol. 2020, 38, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Greve, G.; Zimmer, D.; Bresser, H.; Berberich, B.; Langova, R.; Stomper, J.; Rubarth, A.; Feuerbach, L.; Lipka, D.B.; et al. The Antileukemic Activity of Decitabine upon PML/RARA-Negative AML Blasts Is Supported by All-Trans Retinoic Acid: In Vitro and in Vivo Evidence for Cooperation. Blood Cancer J. 2022, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, Y.; Liang, Z.; Wang, Y.; Chen, F.; Djekidel, M.N.; Li, G.; Zhang, X.; Xiang, S.; Wang, Z.; et al. Alterations of Specific Chromatin Conformation Affect ATRA-Induced Leukemia Cell Differentiation. Cell Death Dis. 2018, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Trus, M.R.; Yang, L.; Suarez Saiz, F.; Bordeleau, L.; Jurisica, I.; Minden, M.D. The Histone Deacetylase Inhibitor Valproic Acid Alters Sensitivity towards All Trans Retinoic Acid in Acute Myeloblastic Leukemia Cells. Leukemia 2005, 19, 1161–1168. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Mere Del Aguila, E.; Tang, X.-H.; Gudas, L.J. Pancreatic Ductal Adenocarcinoma: New Insights into the Actions of Vitamin A. Oncol. Res. Treat. 2022, 45, 291–298. [Google Scholar] [CrossRef]
- Parigiani, M.A.; Mandel, M.; Becker, H.; Medical Center University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany. 2022; unpublished work.
- Carapuça, E.F.; Gemenetzidis, E.; Feig, C.; Bapiro, T.E.; Williams, M.D.; Wilson, A.S.; Delvecchio, F.R.; Arumugam, P.; Grose, R.P.; Lemoine, N.R.; et al. Anti-Stromal Treatment Together with Chemotherapy Targets Multiple Signalling Pathways in Pancreatic Adenocarcinoma. J. Pathol. 2016, 239, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.-H.; Yao, Y.; Kondo, A.; et al. Active Pin1 Is a Key Target of All-Trans Retinoic Acid in Acute Promyelocytic Leukemia and Breast Cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA Mechanically Reprograms Pancreatic Stellate Cells to Suppress Matrix Remodelling and Inhibit Cancer Cell Invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting Pin1 Renders Pancreatic Cancer Eradicable by Synergizing with Immunochemotherapy. Cell 2021, 184, 4753–4771.e27. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I Clinical Trial Repurposing All-Trans Retinoic Acid as a Stromal Targeting Agent for Pancreatic Cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; Coetzee, C.; de Souza, N.; Goulart, M.; Hughes, C.; et al. STAR-PAC: Phase I Clinical Trial Repurposing All Trans Retinoic Acid (ATRA) as Stromal Targeting Agent in a Novel Drug Combination for Pancreatic Cancer. Ann. Oncol. 2019, 30, v267. [Google Scholar] [CrossRef]
- Phase IIb Randomised Clinical Trial Repurposing ATRA as a Stromal Targeting Agent in a Novel Drug Combination for Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04241276 (accessed on 31 August 2022).
- Luu, T.; Frankel, P.; Beumer, J.H.; Lim, D.; Cristea, M.; Appleman, L.J.; Lenz, H.J.; Gandara, D.R.; Kiesel, B.F.; Piekarz, R.L.; et al. Phase I Trial of Belinostat in Combination with 13-Cis-Retinoic Acid in Advanced Solid Tumor Malignancies: A California Cancer Consortium NCI/CTEP Sponsored Trial. Cancer Chemother. Pharmacol. 2019, 84, 1201–1208. [Google Scholar] [CrossRef]
- Tilsed, C.M.; Casey, T.H.; de Jong, E.; Bosco, A.; Zemek, R.M.; Salmons, J.; Wan, G.; Millward, M.J.; Nowak, A.K.; Lake, R.A.; et al. Retinoic Acid Induces an IFN-Driven Inflammatory Tumour Microenvironment, Sensitizing to Immune Checkpoint Therapy. Front. Oncol. 2022, 12, 849793. [Google Scholar] [CrossRef] [PubMed]
- Treatment with Nivolumab and All-Trans Retinoic Acid for Patients with Refractory Pancreatic Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT05482451 (accessed on 31 August 2022).
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET Bromodomain Proteins in Solid Tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [Green Version]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET Bromodomain Inhibitors Block Growth of Pancreatic Cancer Cells in Three-Dimensional Collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Jauset, T.; Massó-Vallés, D.; Martínez-Martín, S.; Beaulieu, M.-E.; Foradada, L.; Fiorentino, F.P.; Yokota, J.; Haendler, B.; Siegel, S.; Whitfield, J.R.; et al. BET Inhibition Is an Effective Approach against KRAS-Driven PDAC and NSCLC. Oncotarget 2018, 9, 18734–18746. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; DeCant, B.T.; Grippo, P.J.; Hwang, R.F.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET Inhibitors Block Pancreatic Stellate Cell Collagen I Production and Attenuate Fibrosis in Vivo. JCI Insight 2017, 2, e88032. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L.; Garcia, P.L.; Fehling, S.C.; Gamblin, T.L.; Vance, R.B.; Council, L.N.; Chen, D.; Yang, E.S.; van Waardenburg, R.C.A.M.; Yoon, K.J. The BET Inhibitor JQ1 Augments the Antitumor Efficacy of Gemcitabine in Preclinical Models of Pancreatic Cancer. Cancers 2021, 13, 3470. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Zeng, L.; van Waardenburg, R.C.A.M.; Yang, E.S.; Yoon, K.J. The BET Inhibitor JQ1 Potentiates the Anticlonogenic Effect of Radiation in Pancreatic Cancer Cells. Front. Oncol. 2022, 12, 925718. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET Inhibitor I-BET762 Inhibits Pancreatic Ductal Adenocarcinoma Cell Proliferation and Enhances the Therapeutic Effect of Gemcitabine. Sci. Rep. 2018, 8, 8102. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; LoRusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N.; Chen, X.; Hu, B.; Freise, K.J.; et al. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Phase IB Trial with OTX015/MK-8628, a Small Molecule Inhibitor of the Bromodomain and Extra-Terminal (BET) Proteins, in Patients with Selected Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02259114 (accessed on 17 August 2022).
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined Inhibition of BET Family Proteins and Histone Deacetylases as a Potential Epigenetics-Based Therapy for Pancreatic Ductal Adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Dong, G.; Li, Y.; Wu, S.; Wang, W.; Sheng, C. Potent Dual BET/HDAC Inhibitors for Efficient Treatment of Pancreatic Cancer. Angew. Chem. Int. Ed. 2020, 59, 3028–3032. [Google Scholar] [CrossRef]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.A.M.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET Inhibitor JQ1 Attenuates Double-Strand Break Repair and Sensitizes Models of Pancreatic Ductal Adenocarcinoma to PARP Inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Phase Ib/II Study of ZEN003694 and Entinostat in Advanced and Refractory Solid Tumors and Lymphomas. Available online: https://clinicaltrials.gov/ct2/show/NCT05053971 (accessed on 17 August 2022).
- Phase 1/2 Safety and Efficacy Study of NUV-868 as Monotherapy and in Combination with Olaparib or Enzalutamide in Adult Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT05252390 (accessed on 17 August 2022).
- Principe, D.R.; Xiong, R.; Li, Y.; Pham, T.N.D.; Kamath, S.D.; Dubrovskyi, O.; Ratia, K.; Huang, F.; Zhao, J.; Shen, Z.; et al. XP-524 Is a Dual-BET/EP300 Inhibitor That Represses Oncogenic KRAS and Potentiates Immune Checkpoint Inhibition in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2116764119. [Google Scholar] [CrossRef]
- Tu, M.; Klein, L.; Espinet, E.; Georgomanolis, T.; Wegwitz, F.; Li, X.; Urbach, L.; Danieli-Mackay, A.; Küffer, S.; Bojarczuk, K.; et al. TNF-α-Producing Macrophages Determine Subtype Identity and Prognosis via AP1 Enhancer Reprogramming in Pancreatic Cancer. Nat. Cancer 2021, 2, 1185–1203. [Google Scholar] [CrossRef]
- Phase I/Ib Trial Evaluating the Safety and Efficacy of BET Inhibitor, ZEN003694 with PD-1 Inhibitor, Nivolumab with or without CTLA-4 Inhibitor, Ipilimumab in Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04840589 (accessed on 17 August 2022).
- A Phase I/IIa Trial with BMS-986158, a Small Molecule Inhibitor of the Bromodomain and Extra-Terminal (BET) Proteins, as Monotherapy or in Combination with Nivolumab in Subjects with Selected Advanced Solid Tumors or Hematologic Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02419417 (accessed on 17 August 2022).
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an Easy Way to Harmonize: A Review of Advances in Clinical Research and Combination Strategies of EZH2 Inhibitors. Clin. Epigenetics 2021, 13, 62. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, W.; Li, B.; Cheng, B.; Lin, K.; Bai, J.; Li, H.; Yang, G. Targeting Ezh2 Could Overcome Docetaxel Resistance in Prostate Cancer Cells. BMC Cancer 2019, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.-J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.-K.; et al. EZH2 Inhibition Sensitizes BRG1 and EGFR Mutant Lung Tumours to TopoII Inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.-H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Z.; Liu, D. Interference with Endogenous EZH2 Reverses the Chemotherapy Drug Resistance in Cervical Cancer Cells Partly by up-Regulating Dicer Expression. Tumour Biol. 2016, 37, 6359–6369. [Google Scholar] [CrossRef]
- Avan, A.; Crea, F.; Paolicchi, E.; Funel, N.; Galvani, E.; Marquez, V.E.; Honeywell, R.J.; Danesi, R.; Peters, G.J.; Giovannetti, E. Molecular Mechanisms Involved in the Synergistic Interaction of the EZH2 Inhibitor 3-Deazaneplanocin A with Gemcitabine in Pancreatic Cancer Cells. Mol. Cancer Ther. 2012, 11, 1735–1746. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Sun, Y.; Liang, L.; Lu, W.; Luo, B.; Wu, Z.; Huo, B.; Hu, Y.; Huang, P.; Wu, Q.; et al. Design and Synthesis of Dual EZH2/BRD4 Inhibitors to Target Solid Tumors. J. Med. Chem. 2022, 65, 6573–6592. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.-M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [Green Version]
- A Phase II Study of Tazemetostat in Solid Tumors Harboring an ARID1A Mutation. Available online: https://clinicaltrials.gov/ct2/show/NCT05023655 (accessed on 17 August 2022).
- An Open-Label, Multicenter, Two-Part, Phase 1 Study to Characterize the Effects of a Moderate CYP3A Inhibitor on the Pharmacokinetics of Tazemetostat (EPZ-6438) (Part A), the Effects of Tazemetostat on the Pharmacokinetics of CYP2C8 and CYP2C19 Substrates, and the Effect of Increased Gastric PH on the Pharmacokinetics of Tazemetostat (Part B) in Subjects with B-Cell Lymphoma or Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03028103 (accessed on 17 August 2022).
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- An Open-Label Phase I/II Study of EZH2 Inhibitor SHR2554 in Combination with Anti-PD-L1/TGFβ Antibody SHR1701 in Patients with Advanced or Metastatic Solid Tumors and Relapsed/Refractory B-Cell Lymphomas. Available online: https://clinicaltrials.gov/ct2/show/NCT04407741 (accessed on 17 August 2022).
- A Phase 1 Study of CPI-1205 with Ipilimumab in Patients with Advanced Solid Tumors Followed by a Phase 2 Basket Study of CPI-1205 with Ipilimumab in Selected Tumor Types Previously Treated with PD-1 or PD-L1 Inhibitors. Available online: https://clinicaltrials.gov/ct2/show/NCT03525795 (accessed on 17 August 2022).
- Combining Epigenetic and Immune Therapy to Beat Cancer. CAIRE Study. Available online: https://clinicaltrials.gov/ct2/show/NCT04705818 (accessed on 17 August 2022).
- Zhou, L.; Mudianto, T.; Ma, X.; Riley, R.; Uppaluri, R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 290–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti-CTLA-4 Therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in Epigenetic Therapeutics with Focus on Solid Tumors. Clin. Epigenetics 2021, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Zopf, S.; Leu, T.; Dietze, O.; Hauser-Kronberger, C.; Hahn, E.G.; Herold, C.; Ocker, M. Apoptosis, Proliferation and Differentiation Patterns Are Influenced by Zebularine and SAHA in Pancreatic Cancer Models. Scand. J. Gastroenterol. 2007, 42, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment Drives Cell State, Plasticity, and Drug Response in Pancreatic Cancer. Cell 2021, 184, 6119–6137.e26. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription Phenotypes of Pancreatic Cancer Are Driven by Genomic Events during Tumor Evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Niemöller, C.; Wehrle, J.; Riba, J.; Claus, R.; Renz, N.; Rhein, J.; Bleul, S.; Stosch, J.M.; Duyster, J.; Plass, C.; et al. Bisulfite-Free Epigenomics and Genomics of Single Cells through Methylation-Sensitive Restriction. Commun. Biol. 2021, 4, 153. [Google Scholar] [CrossRef]
- Gupta, S.; Pramanik, D.; Mukherjee, R.; Campbell, N.R.; Elumalai, S.; de Wilde, R.F.; Hong, S.-M.; Goggins, M.G.; De Jesus-Acosta, A.; Laheru, D.; et al. Molecular Determinants of Retinoic Acid Sensitivity in Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 280–289. [Google Scholar] [CrossRef]
- Azad, N.; Zahnow, C.A.; Rudin, C.M.; Baylin, S.B. The Future of Epigenetic Therapy in Solid Tumours—Lessons from the Past. Nat. Rev. Clin. Oncol. 2013, 10, 256–266. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Lechner, S.; Malgapo, M.I.P.; Grätz, C.; Steimbach, R.R.; Baron, A.; Rüther, P.; Nadal, S.; Stumpf, C.; Loos, C.; Ku, X.; et al. Target Deconvolution of HDAC Pharmacopoeia Reveals MBLAC2 as Common off-Target. Nat. Chem. Biol. 2022, 18, 812–820. [Google Scholar] [CrossRef]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological Rationale for the Use of DNA Methyltransferase Inhibitors as New Strategy for Modulation of Tumor Response to Chemotherapy and Radiation. Mol. Cancer 2010, 9, 305. [Google Scholar] [CrossRef] [Green Version]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent Developments of HDAC Inhibitors: Emerging Indications and Novel Molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET Inhibitors: A Novel Epigenetic Approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a First-in-Class Reversible DNMT1-Selective Inhibitor with Improved Tolerability and Efficacy in Acute Myeloid Leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Porcu, P.; Haverkos, B.; Brem, E.; Vallurupalli, A.; Feldman, T.; Alpdogan, O.; Brammer, J.E.; Bryan, L.J.; Barta, S.K.; Obrzut, S.; et al. A Phase Ib/II Study of Oral Nanatinostat (N) and Valganciclovir (VG) in Subjects with Epstein-Barr Virus (EBV)-Associated Lymphomas. J. Clin. Oncol. 2019, 37, 7551. [Google Scholar] [CrossRef]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus Lenalidomide, and Dexamethasone in Relapsed or Refractory Multiple Myeloma: A Multicentre Phase 1b Trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]

| Test | Type | Sample | Arms | Results | Reference |
|---|---|---|---|---|---|
| Methylation of a 5-gene panel | Diagnostic | Blood | PDAC Healthy controls | Differentiated PDAC from controls; sensitivity 76%, specificity 59% | [81] |
| Methylation of a 6-gene panel | Diagnostic | Blood | PDAC Chronic pancreatitis Healthy controls | Differentiated PDAC from healthy controls but not chronic pancreatitis | [82] |
| Hypermethylation of NPTX2 | Diagnostic | Blood | PDAC Chronic pancreatitis Biliary stone diseases | Differentiated PDAC from chronic pancreatitis; sensitivity 80%, specificity 76% | [83] |
| Hypermethylation of NPTX2 and SPARC | Diagnostic Prognostic | Blood | PDAC Chronic pancreatitis | Differentiated PDAC from chronic pancreatitis Associated with poor survival | [84] |
| Methylation of BNC1 and ADAMTS1 | Diagnostic | Blood | PDAC No PDAC | Differentiated PDAC from controls without PDAC; sensitivity 97.4%, specificity 91.6% | [85] |
| Methylation of BNC1 and ADAMTS1 | Diagnostic | Blood | PDAC Healthy controls | Differentiated PDAC from controls; sensitivity 81%, specificity 85% | [86] |
| Methylation of an 8-gene panel | Diagnostic | Blood | PDAC Chronic/acute pancreatitis No pancreatic disease | Differentiated PDAC from controls; sensitivity 76%, specificity 83% | [87] |
| Tissue-specific DNA methylation markers | Diagnostic | Blood | PDAC Chronic pancreatitis Healthy controls | Differentiated PDAC and pancreatitis from controls | [88] |
| Panel of differentially methylated regions (DMR) | Diagnostic | Blood | PDAC Other gastrointestinal cancers | Differentiated PDAC from other cancers | [89] |
| Methylation of a 13- gene panel + CA19-9 level | Diagnostic | Blood | PDAC Healthy controls | Detected PDAC across all stages compared to controls; at pre-set specificity 97.5%: sensitivity 92%, specificity 92% | [90] |
| Methylation of a 10- gene panel | Diagnostic Monitoring | Blood | PDAC Benign pancreatic cysts | Distinguished between metastatic PDAC and benign cysts; sensitivity 100%, Specificity 95% Decrease in methylation levels upon treatment | [91] |
| Methylation of RUNX3 + CA19-9 level | Diagnostic | Blood | PDAC Benign pancreatic disease Healthy controls | Detected PDAC compared to other arms; sensitivity 85.5%, specificity 93.5% | [92] |
| Methylation of C13orf18, FER1L4 and BMP3 | Diagnostic | Pancreatic juice | PDAC IPMN with high grade dysplasia Benign disease Healthy controls | Distinguished between any stage of PDAC and controls; at pre-set specificity 86%: sensitivity 83% Identified patients with stage I or II PDAC or IPMN; at pre-set specificity 86%: sensitivity 80% | [94] |
| Methylation of TBX15, BMP3 | Diagnostic | Pancreatic cyst fluid | PDAC High grade dysplasia Low or no dysplasia | Distinguished between PDAC and high grade dysplasia from other conditions; sensitivity and specificity above 90% | [95] |
| Methylation of BMP3 and mutant KRAS | Diagnostic | Stool | PDAC Healthy controls | Distinguished between PDAC and controls; at pre-set specificity 90%: sensitivity 67% | [96] |
| Number and specific set of hypermethylated genes | Prognostic | Blood | PDAC | Differentiated between metastatic disease and earlier stages | [97] |
| Number of hypermethylated genes | Prognostic | Blood | PDAC | Patients with more than 10 hypermethylated genes (of 28 analyzed) had worse survival outcomes | [98] |
| Methylation of a predefined gene panel | Prognostic | Blood | PDAC | Overall and disease stage-specific risk models based on the methylation status of the genes analyzed | [98] |
| Methylation of HOXD8 and POU4F1 | Prognostic | Blood | PDAC | Independent prognostic marker for PFS and OS | [100] |
| A panel of nucleosomal biomarkers with or without CA19-9 | Diagnostic | Blood | PDAC Benign pancreatic disease Healthy controls | Two models distinguished PDAC from other arms at pre-set specificity (90%); model 1 (5 nucleosomal biomarkers): sensitivity 72%; model 2 (4 nucleosomal biomarkers + CA19-9): sensitivity 92% | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elrakaybi, A.; Ruess, D.A.; Lübbert, M.; Quante, M.; Becker, H. Epigenetics in Pancreatic Ductal Adenocarcinoma: Impact on Biology and Utilization in Diagnostics and Treatment. Cancers 2022, 14, 5926. https://doi.org/10.3390/cancers14235926
Elrakaybi A, Ruess DA, Lübbert M, Quante M, Becker H. Epigenetics in Pancreatic Ductal Adenocarcinoma: Impact on Biology and Utilization in Diagnostics and Treatment. Cancers. 2022; 14(23):5926. https://doi.org/10.3390/cancers14235926
Chicago/Turabian StyleElrakaybi, Asmaa, Dietrich A. Ruess, Michael Lübbert, Michael Quante, and Heiko Becker. 2022. "Epigenetics in Pancreatic Ductal Adenocarcinoma: Impact on Biology and Utilization in Diagnostics and Treatment" Cancers 14, no. 23: 5926. https://doi.org/10.3390/cancers14235926
APA StyleElrakaybi, A., Ruess, D. A., Lübbert, M., Quante, M., & Becker, H. (2022). Epigenetics in Pancreatic Ductal Adenocarcinoma: Impact on Biology and Utilization in Diagnostics and Treatment. Cancers, 14(23), 5926. https://doi.org/10.3390/cancers14235926