

Development of Allogeneic Stem Cell-Based Platform for Delivery and Potentiation of Oncolytic Virotherapy
 , and
, and 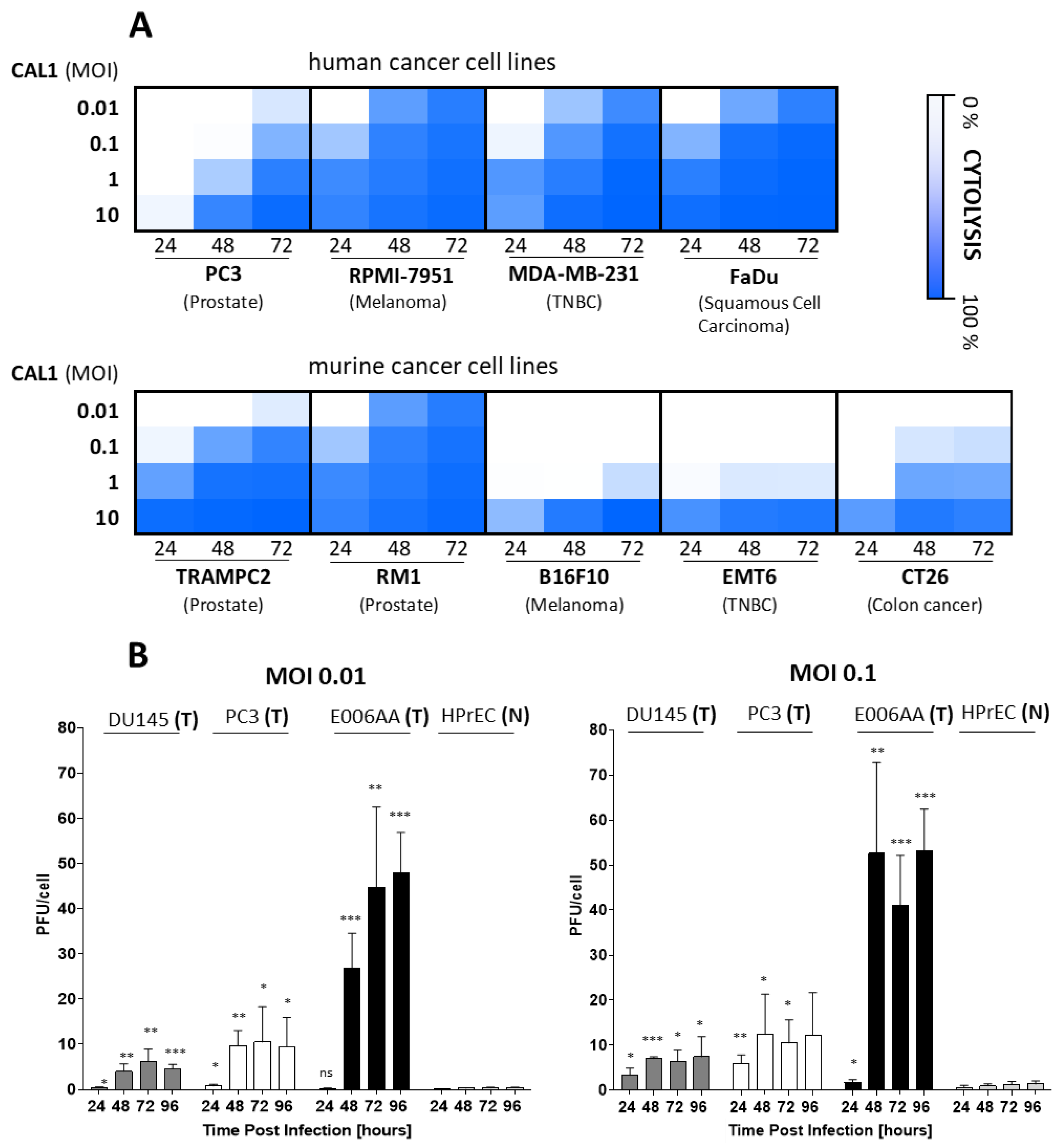{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Next Generation Sequencing (NGS)
2.2. Cell Culture
2.3. Virus Amplification Assay
2.4. Adipose-Derived Stem Cell Generation and Characterization
2.5. SNV1 Production
2.6. Plaque Assay
2.7. Serum Collection
2.8. Vaccinia Virus Neutralizing Antibody Analysis
2.9. Animal Tumor Models
2.10. Real Time Cell Analysis of Virus Mediated Cytolysis
2.11. Guide RNA Target Intergenic Locus (ORF-157 and ORF-158)
2.12. Donor Vector
2.13. Cas9HFc Vector
2.14. Transfection and Viral Infection
2.15. Virus Purification
2.16. PCR Confirmation
2.17. Statistics
3. Results
3.1. CAL1 Vaccinia Virus Derived from ACAM2000™ Is a Potent Oncolytic Virus and Preferentially Amplifies in Cancer Cells
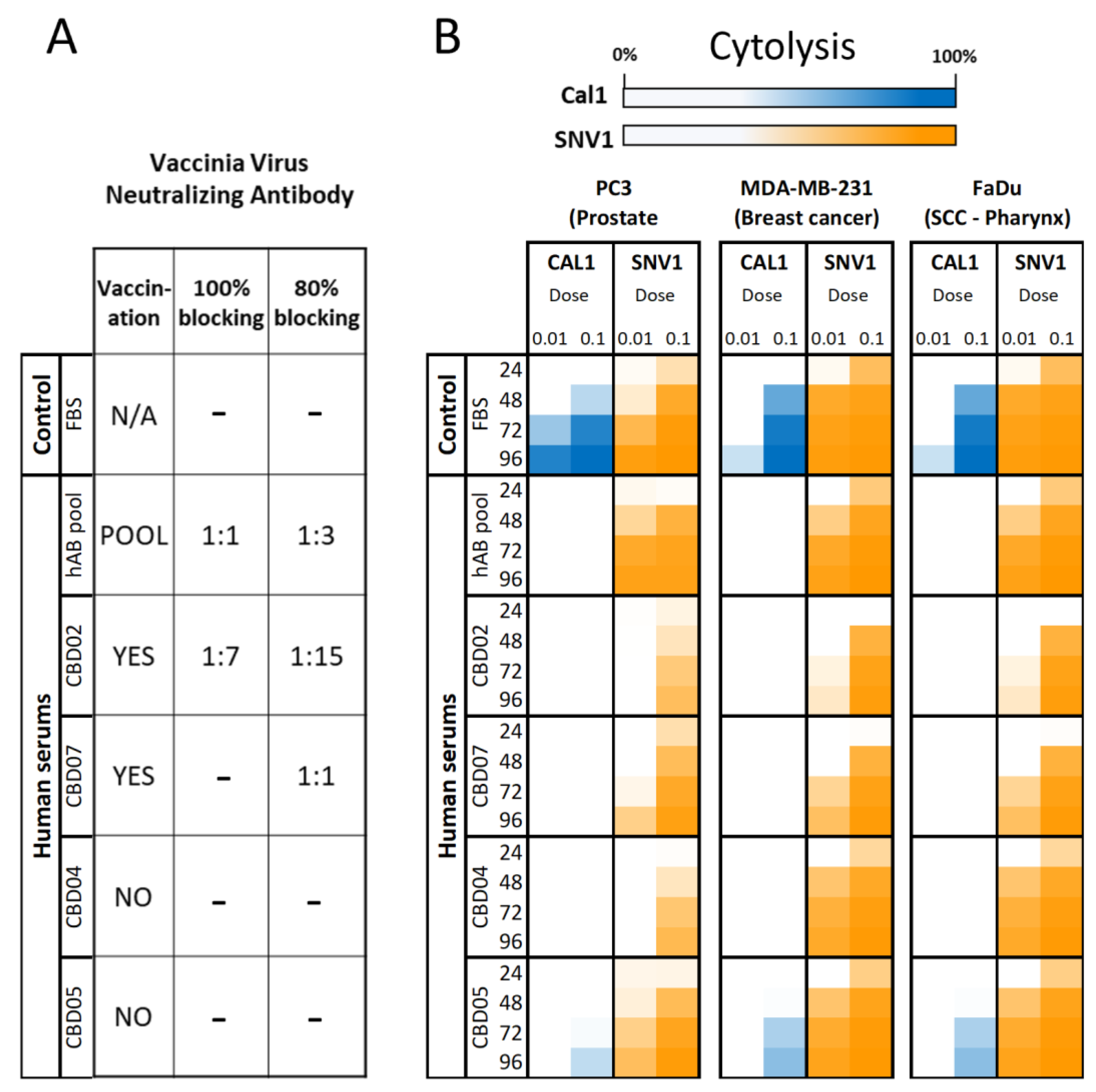
3.2. AD-MSCs Ad-Mscs Protect Oncolytic Viruses from Elimination by the Human Humoral Immunity
3.3. The Use of AD-MSC Significantly Increases the Therapeutic Efficacy of CAL1 in Tumor-Bearing Mouse Models
3.4. CAL1 Vaccinia Virus as a Backbone to Express Therapeutic Genes in Cancer Cells
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minev, B.R.; Lander, E.; Feller, J.F.; Berman, M.; Greenwood, B.M.; Minev, I.; Santidrian, A.F.; Nguyen, D.; Draganov, D.; Killinc, M.O.; et al. First-in-human study of TK-positive oncolytic vaccinia virus delivered by adipose stromal vascular fraction cells. J. Transl. Med. 2019, 17, 271–315. [Google Scholar] [CrossRef]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.-J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [Green Version]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.M.; Nieva, J.; Hwang, T.-H.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 Study of Intratumoral Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus, in Pediatric Cancer Patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.M.B.U.; Hong, Y.M.; Ornella, M.S.C.; Ngabire, D.; Jang, H.; Cho, E.; Kim, E.-K.; Hale, J.J.J.; Kim, C.H.; Ahn, S.C.; et al. Engineering and Preclinical Evaluation of Western Reserve Oncolytic Vaccinia Virus Expressing A167Y Mutant Herpes Simplex Virus Thymidine Kinase. Biomedicines 2020, 8, 426. [Google Scholar] [CrossRef]
- Downs-Canner, S.; Guo, Z.S.; Ravindranathan, R.; Breitbach, C.J.; O’Malley, M.E.; Jones, H.L.; Moon, A.; McCart, J.A.; Shuai, Y.; Zeh, H.J.; et al. Phase 1 Study of Intravenous Oncolytic Poxvirus (vvDD) in Patients With Advanced Solid Cancers. Mol. Ther. 2016, 24, 1492–1501. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, A.; Cadet, V.E.; Hielscher, A. The mechanisms of genetically modified vaccinia viruses for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2015, 95, 407–416. [Google Scholar] [CrossRef]
- Buller, R.M.L.; Smith, G.L.; Cremer, K.; Notkins, A.L.; Moss, B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature 1985, 317, 813–815. [Google Scholar] [CrossRef]
- Belisario, J.C.; Milton, G.W. The experimental local therapy of cutaneous metastases of malignant melanoblastomas with cow pox vaccine or colcemid (demecolcine or omaine). Aust. J. Dermatol. 1961, 6, 113–118. [Google Scholar] [CrossRef]
- Everall, J.; Wand, J.; O’Doherty, C.; Dowd, P. Treatment of primary melanoma by intralesional vaccinia before excision. Lancet 1975, 306, 583–586. [Google Scholar] [CrossRef]
- Hunter-Craig, I.; Newton, K.A.; Westbury, G.; Lacey, B.W. Use of Vaccinia Virus in the Treatment of Metastatic Malignant Melanoma. Br. Med. J. 1970, 2, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Weltzin, R.; Liu, J.; Pugachev, K.V.; Myers, G.A.; Coughlin, B.; Blum, P.S.; Nichols, R.; Johnson, C.; Cruz, J.; Kennedy, J.S.; et al. Clonal vaccinia virus grown in cell culture as a new smallpox vaccine. Nat. Med. 2003, 9, 1125–1130. [Google Scholar] [CrossRef]
- Monath, T.P.; Caldwell, J.R.; Mundt, W.; Fusco, J.; Johnson, C.S.; Buller, M.; Liu, J.; Gardner, B.; Downing, G.; Blum, P.S.; et al. ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain)—A second-generation smallpox vaccine for biological defense. Int. J. Infect. Dis. 2004, 8, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.D.; Da Silva, M.; Frace, A.M.; Sammons, S.A.; Olsen-Rasmussen, M.; Upton, C.; Buller, R.M.L.; Chen, N.; Feng, Z.; Roper, R.L.; et al. Genomic differences of Vaccinia virus clones from Dryvax smallpox vaccine: The Dryvax-like ACAM2000 and the mouse neurovirulent Clone-3. Vaccine 2007, 25, 8807–8832. [Google Scholar] [CrossRef]
- Frey, S.E.; Newman, F.K.; Kennedy, J.S.; Ennis, F.; Abate, G.; Hoft, D.F.; Monath, T.P. Comparison of the safety and immunogenicity of ACAM1000, ACAM2000 and Dryvax® in healthy vaccinia-naive adults. Vaccine 2009, 27, 1637–1644. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Symons, J.A.; Alcamí, A.; Smith, G.L. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 1995, 81, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 129, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef]
- Hadryś, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef]
- Mooney, R.; Majid, A.A.; Batalla-Covello, J.; Machado, D.; Liu, X.; Gonzaga, J.; Tirughana, R.; Hammad, M.; Dellinger, T.; Lesniak, M.S.; et al. Enhanced Delivery of Oncolytic Adenovirus by Neural Stem Cells for Treatment of Metastatic Ovarian Cancer. Mol. Ther. Oncolytics 2018, 12, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zendedel, E.; Atkin, S.L.; Sahebkar, A. Use of stem cells as carriers of oncolytic viruses for cancer treatment. J. Cell. Physiol. 2019, 234, 14906–14913. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, M.O.; Santidrian, A.; Minev, I.; Toth, R.; Draganov, D.; Nguyen, D.; Lander, E.; Berman, M.; Minev, B.; Szalay, A.A. The ratio of ADSCs to HSC-progenitors in adipose tissue derived SVF may provide the key to predict the outcome of stem-cell therapy. Clin. Transl. Med. 2018, 7, 5. [Google Scholar] [CrossRef]
- Draganov, D.D.; Santidrian, A.F.; Minev, I.; Nguyen, D.; Kilinc, M.O.; Petrov, I.; Vyalkova, A.; Lander, E.; Berman, M.; Minev, B.; et al. Delivery of oncolytic vaccinia virus by matched allogeneic stem cells overcomes critical innate and adaptive immune barriers. J. Transl. Med. 2019, 17, 100. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Tselikas, L.; de Baere, T.; Houot, R. Intratumoral immunotherapy: Using the tumor as the remedy. Ann. Oncol. 2017, 28, xii33–xii43. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A.; International Society for Cellular, T. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Evgin, L.; Acuna, S.A.; De Souza, C.T.; Marguerie, M.; Lemay, C.G.; Ilkow, C.S.; Findlay, C.S.; Falls, T.; Parato, K.A.; Hanwell, D.; et al. Complement Inhibition Prevents Oncolytic Vaccinia Virus Neutralization in Immune Humans and Cynomolgus Macaques. Mol. Ther. 2015, 23, 1066–1076. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Zhang, W.; He, J.; Liu, Y.; Song, Z.; Zhou, Z.; Zheng, M.; Hu, Y. Novel, real-time cell analysis for measuring viral cytopathogenesis and the efficacy of neutralizing antibodies to the 2009 influenza A (H1N1) virus. PLoS ONE 2012, 7, e31965. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liang, C.; Yu, Y.A.; Chen, N.; Dandekar, T.; Szalay, A.A. The highly attenuated oncolytic recombinant vaccinia virus GLV-1h68: Comparative genomic features and the contribution of F14.5L inactivation. Mol. Genet. Genom. 2009, 282, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Burdick, K.H. Malignant Melanoma Treated with Vaccinia Injections. Arch. Dermatol. 1960, 82, 189. [Google Scholar]
- Burdick, K.H.; Hawk, W.A. Vitiligo in a case of vaccinia virus-treated melanoma. Cancer 1964, 17, 708–712. [Google Scholar] [CrossRef]
- Hansen, R.M.; Libnoch, J.A. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch. Intern. Med. 1978, 138, 1137–1138. [Google Scholar] [CrossRef]
- Yettra, M. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch. Intern. Med. 1979, 139, 603. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Maguire, H.C.; McCue, P.; Lee, S.S.; Alexander, A.; Nazarian, L.N.; Eisenlohr, L.C.; Nathan, F.; Berd, D.; Lattime, E.C. A pilot study demonstrating the feasibility of using intratumoral vaccinia injections as a vector for gene transfer. Vaccine Res. 1995, 4, 55–69. [Google Scholar]
- Nalca, A.; Zumbrun, E. ACAM2000™: The new smallpox vaccine for United States Strategic National Stockpile. Drug Des. Dev. Ther. 2010, 4, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chen, N.; Feng, Z.; Buller, R.M.L.; Osborne, J.; Harms, T.; Damon, I.; Upton, C.; Esteban, D.J. Genomic sequence and analysis of a vaccinia virus isolate from a patient with a smallpox vaccine-related complication. Virol. J. 2006, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, C.A.; Earl, P.L.; Wyatt, L.S.; Moss, B. Preparation of Cell Cultures and Vaccinia Virus Stocks. Curr. Protoc. Mol. Biol. 1998, 43, 16. [Google Scholar] [CrossRef]
- Spriggs, M.K.; Hruby, D.E.; Maliszewski, C.R.; Pickup, D.J.; Sims, J.E.; Buller, R.L.; VanSlyke, J. Vaccinia and cowpox viruses encode a novel secreted interleukin-1-binding protein. Cell 1992, 71, 145–152. [Google Scholar] [CrossRef]
- Ong, G.L.; Mattes, M. Mouse strains with typical mammalian levels of complement activity. J. Immunol. Methods 1989, 125, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Ratelade, J.; Verkman, A. Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol. Immunol. 2014, 62, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Chiu, S.M.; Motan, D.A.L.; Zhang, Z.; Chen, L.; Ji, H.-L.; Tse, H.-F.; Fu, Q.-L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [Green Version]
- Moreno, R.; Fajardo, C.A.; Farrera-Sal, M.; Perisé-Barrios, A.J.; Morales-Molina, A.; Al-Zaher, A.A.; García-Castro, J.; Alemany, R. Enhanced Antitumor Efficacy of Oncolytic Adenovirus–loaded Menstrual Blood–derived Mesenchymal Stem Cells in Combination with Peripheral Blood Mononuclear Cells. Mol. Cancer Ther. 2019, 18, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, Z.; Xu, X.; Xu, Z.; Wang, S.; Huang, D.; Li, Y.; Mou, X.; Liu, F.; Xiang, C. Menstrual Blood-Derived Stem Cells as Delivery Vehicles for Oncolytic Adenovirus Virotherapy for Colorectal Cancer. Stem Cells Dev. 2019, 28, 882–896. [Google Scholar] [CrossRef]
- Alfano, A.L.; Candia, A.N.; Cuneo, N.; Guttlein, L.N.; Soderini, A.; Rotondaro, C.; Sganga, L.; Podhajcer, O.L.; Lopez, M.V. Oncolytic Adenovirus-Loaded Menstrual Blood Stem Cells Overcome the Blockade of Viral Activity Exerted by Ovarian Cancer Ascites. Mol. Ther. Oncolytics 2017, 6, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, Y.; Li, M.; Dellinger, T.H.; Mooney, R.; Rahman, M.M.; McFadden, G.; Aboody, K.S.; Hammad, M. NSCs are permissive to oncolytic Myxoma virus and provide a delivery method for targeted ovarian cancer therapy. Oncotarget 2020, 11, 4693–4698. [Google Scholar] [CrossRef]
- Aboody, K.S.; Najbauer, J.; Metz, M.Z.; D’Apuzzo, M.; Gutova, M.; Annala, A.J.; Synold, T.W.; Couture, L.A.; Blanchard, S.; Moats, R.A.; et al. Neural Stem Cell–Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Sci. Transl. Med. 2013, 5, 184ra59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrigan, B.C.P.; Shimizu, Y.; Andreeff, M.; Lang, F.F. Mesenchymal stromal cells for the delivery of oncolytic viruses in gliomas. Cytotherapy 2017, 19, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.X.; Haebe, S.; Lee, A.S.; Westphalen, C.B.; Norton, J.A.; Jiang, W.; Levy, R. Intratumoral Immunotherapy for Early-stage Solid Tumors. Clin. Cancer Res. 2020, 26, 3091–3099. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, D.H.; Herrmann, T.; Härtl, B.; Draganov, D.; Minev, I.; Neuharth, F.; Gomez, A.; Alamillo, A.; Schneider, L.E.; Kleinholz, D.; et al. Development of Allogeneic Stem Cell-Based Platform for Delivery and Potentiation of Oncolytic Virotherapy. Cancers 2022, 14, 6136. https://doi.org/10.3390/cancers14246136
Nguyen DH, Herrmann T, Härtl B, Draganov D, Minev I, Neuharth F, Gomez A, Alamillo A, Schneider LE, Kleinholz D, et al. Development of Allogeneic Stem Cell-Based Platform for Delivery and Potentiation of Oncolytic Virotherapy. Cancers. 2022; 14(24):6136. https://doi.org/10.3390/cancers14246136
Chicago/Turabian StyleNguyen, Duong Hoang, Thomas Herrmann, Barbara Härtl, Dobrin Draganov, Ivelina Minev, Forrest Neuharth, Alberto Gomez, Ashley Alamillo, Laura Edith Schneider, Daniela Kleinholz, and et al. 2022. "Development of Allogeneic Stem Cell-Based Platform for Delivery and Potentiation of Oncolytic Virotherapy" Cancers 14, no. 24: 6136. https://doi.org/10.3390/cancers14246136
APA StyleNguyen, D. H., Herrmann, T., Härtl, B., Draganov, D., Minev, I., Neuharth, F., Gomez, A., Alamillo, A., Schneider, L. E., Kleinholz, D., Minev, B., & Santidrian, A. F. (2022). Development of Allogeneic Stem Cell-Based Platform for Delivery and Potentiation of Oncolytic Virotherapy. Cancers, 14(24), 6136. https://doi.org/10.3390/cancers14246136