

Antagonistic Functions of Androgen Receptor and NF-κB in Prostate Cancer—Experimental and Computational Analyses

 , , ,
, , , 

Abstract
:Simple Summary
Abstract
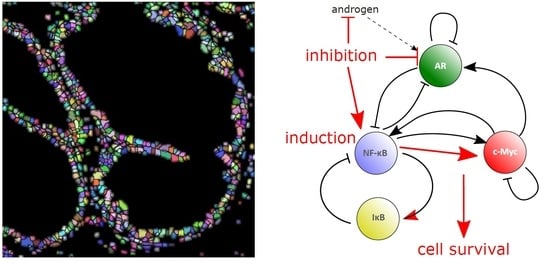
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Materials
2.2. ABCD Assay
2.3. Reporter Gene Assays
2.4. Western Blot
2.5. Staining of Mouse Prostate Tissue for AR and p65-NF-κB
2.6. Transcriptomics Analysis
2.7. Ingenuity Pathway Analysis (IPA)
2.8. Gene Set Enrichment Analysis and Overrepresentation Analysis
2.9. NetworkAnalyst Approach to Study Enriched Pathways
2.10. Mathematical Simulation of a Dynamic Network of AR, NF-κB, IκB, and c-Myc
2.11. Statistics
3. Results
3.1. Activation of the Androgen Receptor Pathway Leads to a Downregulation of NF-κB Signaling
3.2. Treatment of LNCaP and VCaP Cells with Antiandrogens Leads to an Upregulation of NF-κB Signaling and a Reduction of Cell Death
3.3. Mutual Inhibition of AR and NF-κB Signaling Is Also Evident In Vivo
3.4. Long-Term Androgen Deprivation Upregulates NF-κB Activity and Inflammatory Pathways
3.5. Loss of PTEN Upregulates NF-κB Signaling as Well
3.6. Mathematical Model of a Dynamic AR/NF-κB/c-Myc Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Laversanne, M.; Ervik, M.; Lam, F.W.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow. Lyon, France: International Agency for Research on Cancer. Available online: https://gco.iarc.fr/tomorrow (accessed on 23 February 2022).
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Kyprianou, N.; Isaacs, J.T. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology 1988, 122, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, N.; Wikstrom, P.; Stattin, P.; Bergh, A. Cell proliferation and apoptosis in prostate tumors and adjacent non-malignant prostate tissue in patients at different time-points after castration treatment. Prostate 2005, 62, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, N.; Bergh, A.; Stattin, P.; Wikstrom, P. Castration-induced epithelial cell death in human prostate tissue is related to locally reduced IGF-1 levels. Prostate 2007, 67, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Antony, L.; van der Schoor, F.; Dalrymple, S.L.; Isaacs, J.T. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate 2014, 74, 1118–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Jiang, M.; Grabowska, M.M.; Li, J.; Connelly, Z.M.; Zhang, J.; Hayward, S.W.; Cates, J.M.; Han, G.; Yu, X. Androgen receptor differentially regulates the proliferation of prostatic epithelial cells in vitro and in vivo. Oncotarget 2016, 7, 70404–70419. [Google Scholar] [CrossRef] [Green Version]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Debes, J.D.; Tindall, D.J. Mechanisms of Androgen-Refractory Prostate Cancer. N. Engl. J. Med. 2004, 351, 1488–1490. [Google Scholar] [CrossRef]
- Mussbacher, M.; Schossleitner, K.; Kral-Pointner, J.B.; Salzmann, M.; Schrammel, A.; Schmid, J.A. More than Just a Monolayer: The Multifaceted Role of Endothelial Cells in the Pathophysiology of Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 483–492. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, J.A.; Birbach, A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)--a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008, 19, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Altuwaijri, S.; Lin, H.K.; Chuang, K.H.; Lin, W.J.; Yeh, S.; Hanchett, L.A.; Rahman, M.M.; Kang, H.Y.; Tsai, M.Y.; Zhang, Y.; et al. Interruption of nuclear factor kappaB signaling by the androgen receptor facilitates 12-O-tetradecanoylphorbolacetate-induced apoptosis in androgen-sensitive prostate cancer LNCaP cells. Cancer Res. 2003, 63, 7106–7112. [Google Scholar]
- Keller, E.T.; Chang, C.; Ershler, W.B. Inhibition of NFkappaB activity through maintenance of IkappaBalpha levels contributes to dihydrotestosterone-mediated repression of the interleukin-6 promoter. J. Biol. Chem. 1996, 271, 26267–26275. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.D.; Sawyers, C.L. NF-kappa B activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol. Cell. Biol. 2002, 22, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Gasparian, A.V.; Yao, Y.J.; Kowalczyk, D.; Lyakh, L.A.; Karseladze, A.; Slaga, T.J.; Budunova, I.V. The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell Sci. 2002, 115, 141–151. [Google Scholar] [CrossRef]
- Nelius, T.; Filleur, S.; Yemelyanov, A.; Budunova, I.; Shroff, E.; Mirochnik, Y.; Aurora, A.; Veliceasa, D.; Xiao, W.; Wang, Z.; et al. Androgen receptor targets NFkappaB and TSP1 to suppress prostate tumor growth in vivo. Int. J. Cancer 2007, 121, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Altuwaijri, S.; Deng, F.; Chen, L.; Lal, P.; Bhanot, U.K.; Korets, R.; Wenske, S.; Lilja, H.G.; Chang, C.; et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 2009, 175, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Austin, D.C.; Strand, D.W.; Love, H.L.; Franco, O.E.; Jang, A.; Grabowska, M.M.; Miller, N.L.; Hameed, O.; Clark, P.E.; Fowke, J.H.; et al. NF-κB and androgen receptor variant expression correlate with human BPH progression. Prostate 2016, 76, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Duyao, M.P.; Buckler, A.J.; Sonenshein, G.E. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc. Natl. Acad. Sci. USA 1990, 87, 4727–4731. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Hochreiter, B.; Basílio, J.; Gleitsmann, V.; Panhuber, A.; Pardo-Garcia, A.; Hoesel, B.; Salzmann, M.; Resch, U.; Noreen, M.; et al. The inflammatory kinase IKKα phosphorylates and stabilizes c-Myc and enhances its activity. Mol. Cancer 2021, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Abu-Baker, S.; Joshi, J.; Galvez, A.; Castilla, E.A.; Canamero, M.; Collado, M.; Saez, C.; Moreno-Bueno, G.; Palacios, J.; et al. Simultaneous inactivation of Par-4 and PTEN in vivo leads to synergistic NF-{kappa}B activation and invasive prostate carcinoma. Proc. Natl. Acad. Sci. USA 2009, 106, 12962–12967. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Malkani, N.; Hochreiter, B.; Basílio, J.; Sughra, K.; Ilyas, M.; Schmid, J.A. Sequence-function correlations and dynamics of ERG isoforms. ERG8 is the black sheep of the family. Biochim. Biophys. Acta 2016, 1863, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sughra, K.; Birbach, A.; de Martin, R.; Schmid, J.A. Interaction of the TNFR-receptor associated factor TRAF1 with I-kappa B kinase-2 and TRAF2 indicates a regulatory function for NF-kappa B signaling. PLoS ONE 2010, 5, e12683. [Google Scholar] [CrossRef]
- D’Antonio, J.M.; Ma, C.; Monzon, F.A.; Pflug, B.R. Longitudinal analysis of androgen deprivation of prostate cancer cells identifies pathways to androgen independence. Prostate 2008, 68, 698–714. [Google Scholar] [CrossRef]
- Wang, X.D.; Wang, B.E.; Soriano, R.; Zha, J.; Zhang, Z.; Modrusan, Z.; Cunha, G.R.; Gao, W.Q. Expression profiling of the mouse prostate after castration and hormone replacement: Implication of H-cadherin in prostate tumorigenesis. Differentiation 2007, 75, 219–234. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Birbach, A.; Eisenbarth, D.; Kozakowski, N.; Ladenhauf, E.; Schmidt-Supprian, M.; Schmid, J.A. Persistent inflammation leads to proliferative neoplasia and loss of smooth muscle cells in a prostate tumor model. Neoplasia 2011, 13, 692–703. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- R Core Team. R Foundation for Statistical Computing R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 16 August 2022).
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Mark Dunning, A.L. illuminaHumanv4.db. Bioconductor 2017. [Google Scholar] [CrossRef]
- Kauffmann, A.; Rayner, T.F.; Parkinson, H.; Kapushesky, M.; Lukk, M.; Brazma, A.; Huber, W. Importing ArrayExpress datasets into R/Bioconductor. Bioinformatics 2009, 25, 2092–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.; Carey, V.J.; Huber, W.; Hahne, F. genefilter: Genefilter: Methods for filtering genes from high-throughput experiments. R package version 1.78.0. Available online: https://bioconductor.org/packages/release/bioc/html/genefilter.html (accessed on 16 August 2022).
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2013, 30, 523–530. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Zhang, X.; Guan, N.; Bo, X.; Barnes, M.R.; Luo, Z. Gene Ranking of RNA-Seq Data via Discriminant Non-Negative Matrix Factorization. PLoS ONE 2015, 10, e0137782. [Google Scholar] [CrossRef]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Hanel, R.; Pöchacker, M.; Schölling, M.; Thurner, S. A self-organized model for cell-differentiation based on variations of molecular decay rates. PLoS ONE 2012, 7, e36679. [Google Scholar] [CrossRef]
- Hanel, R.; Pöchacker, M.; Thurner, S. Living on the edge of chaos: Minimally nonlinear models of genetic regulatory dynamics. Philos. Trans. A Math. Phys. Eng. Sci. 2010, 368, 5583–5596. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2—Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 16 August 2022).
- Hu, L.; Chen, X.; Narwade, N.; Lim, M.G.L.; Chen, Z.; Tennakoon, C.; Guan, P.; Chan, U.I.; Zhao, Z.; Deng, M.; et al. Single-cell analysis reveals androgen receptor regulates the ER-to-Golgi trafficking pathway with CREB3L2 to drive prostate cancer progression. Oncogene 2021, 40, 6479–6493. [Google Scholar] [CrossRef]
- Johnston, T.J.; Shaw, G.L.; Lamb, A.D.; Parashar, D.; Greenberg, D.; Xiong, T.; Edwards, A.L.; Gnanapragasam, V.; Holding, P.; Herbert, P.; et al. Mortality Among Men with Advanced Prostate Cancer Excluded from the ProtecT Trial. Eur. Urol. 2017, 71, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. Loss of PTEN Expression Induces NF-kB Via PI3K/Akt Pathway Involving Resistance to Chemotherapy in Acute Lymphoblastic Leukemia Cell Lines. Blood 2004, 104, 4438. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, S.; Li, L.; Kemp, C.J.; Jiang, C.; Lü, J. Proteomic and transcriptomic profiling of Pten gene-knockout mouse model of prostate cancer. Prostate 2020, 80, 588–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokic, D.; Hanel, R.; Thurner, S. A fast and efficient gene-network reconstruction method from multiple over-expression experiments. BMC Bioinform. 2009, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuchbery, R.; Macintyre, G.; Cmero, M.; Harewood, L.M.; Peters, J.S.; Costello, A.J.; Hovens, C.M.; Corcoran, N.M. Reduction in expression of the benign AR transcriptome is a hallmark of localised prostate cancer progression. Oncotarget 2016, 7, 31384–31392. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Severson, T.; Qiu, X.; Alshalalfa, M.; Sjostrom, M.; Quigley, D.; Bergman, A.; Long, H.; Feng, F.; Freedman, M.L.; Zwart, W.; et al. Androgen receptor reprogramming demarcates prognostic, context-dependent gene sets in primary and metastatic prostate cancer. Clin. Epigenet. 2022, 14, 60. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basílio, J.; Hochreiter, B.; Hoesel, B.; Sheshori, E.; Mussbacher, M.; Hanel, R.; Schmid, J.A. Antagonistic Functions of Androgen Receptor and NF-κB in Prostate Cancer—Experimental and Computational Analyses. Cancers 2022, 14, 6164. https://doi.org/10.3390/cancers14246164
Basílio J, Hochreiter B, Hoesel B, Sheshori E, Mussbacher M, Hanel R, Schmid JA. Antagonistic Functions of Androgen Receptor and NF-κB in Prostate Cancer—Experimental and Computational Analyses. Cancers. 2022; 14(24):6164. https://doi.org/10.3390/cancers14246164
Chicago/Turabian StyleBasílio, José, Bernhard Hochreiter, Bastian Hoesel, Emira Sheshori, Marion Mussbacher, Rudolf Hanel, and Johannes A. Schmid. 2022. "Antagonistic Functions of Androgen Receptor and NF-κB in Prostate Cancer—Experimental and Computational Analyses" Cancers 14, no. 24: 6164. https://doi.org/10.3390/cancers14246164
APA StyleBasílio, J., Hochreiter, B., Hoesel, B., Sheshori, E., Mussbacher, M., Hanel, R., & Schmid, J. A. (2022). Antagonistic Functions of Androgen Receptor and NF-κB in Prostate Cancer—Experimental and Computational Analyses. Cancers, 14(24), 6164. https://doi.org/10.3390/cancers14246164